Naujos publikacijos
Nauji atradimai padeda geriau suprasti Retto sindromo priežastis
Paskutinį kartą peržiūrėta: 02.07.2025

Visas „iLive“ turinys yra peržiūrėtas medicinoje arba tikrinamas, kad būtų užtikrintas kuo didesnis faktinis tikslumas.
Mes turime griežtas įsigijimo gaires ir susiejamos tik su geros reputacijos žiniasklaidos svetainėmis, akademinių tyrimų institucijomis ir, jei įmanoma, medicininiu požiūriu peržiūrimais tyrimais. Atkreipkite dėmesį, kad skliausteliuose ([1], [2] ir tt) esantys numeriai yra paspaudžiami nuorodos į šias studijas.
Jei manote, kad bet koks mūsų turinys yra netikslus, pasenęs arba kitaip abejotinas, pasirinkite jį ir paspauskite Ctrl + Enter.

Retto sindromas yra retas neurologinės raidos sutrikimas, kuriam šiuo metu nėra išgydymo ar gero gydymo. Jis sukelia sunkius fizinius ir kognityvinius simptomus, iš kurių daugelis sutampa su autizmo spektro sutrikimais.
Retto sindromą sukelia MECP2 geno mutacijos, kuris yra labai ekspresuojamas smegenyse ir, atrodo, atlieka svarbų vaidmenį palaikant neuronų sveikatą. Šis genas yra X chromosomoje, ir sindromas daugiausia paveikia mergaites. Norėdami sukurti Retto sindromo gydymo būdus, tyrėjai nori geriau suprasti MECP2 ir jo funkcijas smegenyse.
Tyrėjai, įskaitant Whitehead instituto bendraįkūrėją Rudolfą Jaenischą, MECP2 geną tyrinėjo jau dešimtmečius, tačiau daugelis pagrindinių faktų apie šį geną liko nežinomi. Geno koduojamas baltymas MECP2 dalyvauja genų reguliavime; jis jungiasi prie DNR ir daro įtaką įvairių kitų genų raiškos lygiams arba jų gaminamo baltymo kiekiui.
Tačiau tyrėjai neturėjo išsamaus MECP2 paveiktų genų sąrašo ir nebuvo sutarimo, kaip MECP2 veikia šiuos genus.
Ankstyvieji MECP2 tyrimai leido manyti, kad jis yra represorius, mažinantis savo taikinių genų ekspresiją, tačiau Jaenisch ir kitų atlikti tyrimai anksčiau parodė, kad MECP2 taip pat veikia kaip aktyvatorius, didindamas savo taikinių ekspresiją – ir kad jis iš viso gali būti aktyvatorius. Taip pat nebuvo žinomas MECP2 veikimo mechanizmas arba kaip tiksliai šis baltymas sukelia genų ekspresijos pokyčius.
Technologijų apribojimai neleido tyrėjams gauti aiškumo į šiuos klausimus. Tačiau Yanishas, jo laboratorijos podoktorantė Yi Liu ir buvęs Yanisho laboratorijos narys Anthony Flamier, dabar dirbantis Monrealio universiteto CHU Sainte-Justine tyrimų centro docentu, pasitelkė pažangiausius metodus, kad atsakytų į šiuos likusius klausimus apie MECP2 ir įgytų naujų įžvalgų apie jo vaidmenį smegenų sveikatai ir ligoms.
Jų rezultatai buvo paskelbti žurnale „Neuron“, o tyrėjai taip pat sukūrė internetinę savo MECP2 duomenų saugyklą – MECP2-NeuroAtlas portalą – kaip išteklių kitiems tyrėjams.
„Manau, kad šis straipsnis iš esmės pakeis žmonių supratimą apie tai, kaip MECP2 sukelia Retto sindromą. Mes visiškai naujai suprantame mechanizmą ir tai gali atverti naujų galimybių kuriant šios ligos gydymo būdus“, – sako Janisch, kuris taip pat yra MIT biologijos profesorius.
Gilesnis MECP2 supratimas smegenyse
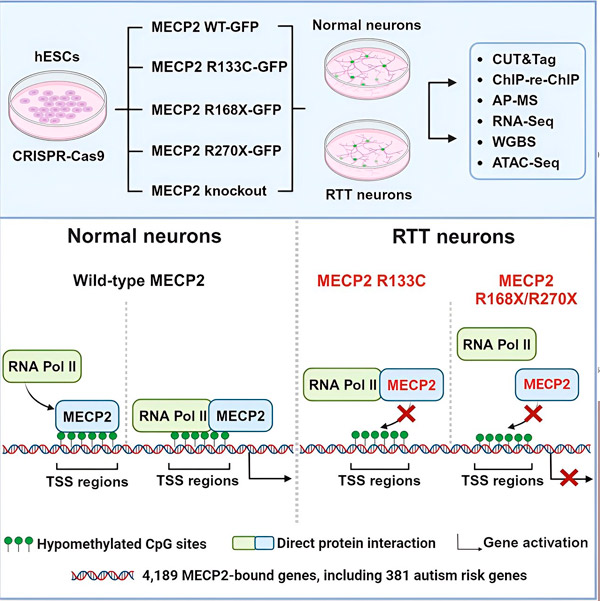
Pirmiausia tyrėjai sukūrė išsamų žemėlapį, kuriame pavaizduota, kur MECP2 jungiasi žmogaus neuronų genų sekose – arba genų viduje, arba šalia jų esančiuose DNR reguliavimo regionuose. Jie naudojo metodą, vadinamą CUT&Tag, kuris gali labai tiksliai nustatyti baltymų sąveiką su DNR.
Tyrėjai rado daugiau nei 4000 su MECP2 susijusių genų. Jie pakartojo savo kartografavimą neuronuose, kuriuose yra dažnos MECP2 mutacijos, susijusios su Retto sindromu, kad nustatytų, kur ligos būsenoje yra sumažėjęs MECP2 kiekis.
Žinant, prie kurių genų jungiasi MECP2, Liu ir Flamier pradėjo nustatyti ryšius tarp MECP2 taikinių ir smegenų sveikatos. Jie nustatė, kad daugelis jo taikinių yra susiję su neuronų aksonų ir sinapsių vystymusi ir funkcija.
Jie taip pat palygino savo MECP2 taikinių sąrašą su Simonso fondo autizmo tyrimų iniciatyvos (SFARI) autizmo sukeltų genų duomenų baze ir nustatė, kad 381 genas toje duomenų bazėje yra MECP2 taikiniai.
Šaltinis: „Neuron“ (2024). DOI: 10.1016/j.neuron.2024.04.007
Šie duomenys gali padėti išsiaiškinti Retto sindromo autizmo simptomų mechanizmus ir būti geras atspirties taškas tiriant galimą MECP2 vaidmenį autizmo atveju.
„Sukūrėme pirmąjį integruotą MECP2 epigenomo žemėlapį sveikatos ir ligų srityse, ir šis žemėlapis gali padėti atlikti būsimus tyrimus“, – sako Liu. „Žinojimas, kurie genai yra MECP2 taikiniai ir kurie genai yra tiesiogiai sutrikdyti ligos metu, suteikia tvirtą pagrindą suprasti Retto sindromą ir užduoti klausimus apie genų reguliavimą neuronuose.“
Tyrėjai taip pat tyrė, ar MECP2 padidino, ar sumažino savo tikslinių genų ekspresiją. Remiantis tuo, kad MECP2 istoriją vieni identifikavo kaip aktyvatorių, o kiti – kaip represorių, Liu ir Flamier rado pavyzdžių, kai MECP2 atliko abu vaidmenis.
Vis dėlto, nors MECP2 dažniau laikomas represoriumi, Liu ir Flamier nustatė, kad jis dažniausiai yra aktyvatorius – tai patvirtina ankstesnius Jaenisch ir Liu tyrimus. Vienas naujas eksperimentas parodė, kad MECP2 aktyvuoja mažiausiai 80 % savo taikinių, o kitas – kad jis aktyvuoja iki 88 % savo taikinių.
Tyrėjų sukurtas tikslinių genų žemėlapis suteikė papildomos informacijos apie MECP2, kaip aktyvatoriaus, vaidmenį. Jie nustatė, kad genai, kuriuos aktyvuoja MECP2, paprastai jungiasi prie DNR srities, esančios prieš geną, vadinamos transkripcijos pradžios vieta.
Tai vieta, kur ląstelės mechanizmai inicijuoja geno transkribavimo į RNR procesą, po kurio RNR transliuojama į funkcinį baltymą, kuris yra genų ekspresijos produktas. MECP2 buvimas transkripcijos pradžios vietoje, kur prasideda genų raiška, atitinka jo, kaip genų aktyvatoriaus, vaidmenį.
Tuomet tyrėjai ėmėsi nustatyti MECP2 vaidmenį genų aktyvavime. Jie ištyrė, prie kokių molekulių, be DNR, MECP2 jungiasi šioje vietoje, ir nustatė, kad MECP2 tiesiogiai sąveikauja su baltymų kompleksu, vadinamu RNR polimeraze II (RNR Pol II). RNR Pol II yra pagrindinė ląstelės mašina, kuri transkribuoja DNR į RNR. RNR Pol II pati negali rasti genų, todėl jai reikia įvairių kofaktorių arba baltymų bendradarbių, kad atliktų savo darbą.
Tyrėjai teigia, kad MECP2 yra vienas iš tokių kofaktorių, padedantis RNR Pol II inicijuoti transkripciją genuose, prie kurių jungiasi MECP2. MECP2 struktūrinė analizė nustatė molekulės dalis, kurios jungiasi prie RNR Pol II, o kiti eksperimentai patvirtino, kad MECP2 praradimas sumažina RNR Pol II buvimą atitinkamose transkripcijos pradžios vietose, taip pat tikslinių genų ekspresijos lygius.
Tai rodo, kad Retto sindromą gali sukelti sumažėjusi MECP2 taikinių genų transkripcija dėl MECP2 mutacijų, kurios neleidžia jam prisijungti prie RNR Pol II arba prie DNR. Remiantis šia idėja, dažniausios su liga susijusios MECP2 mutacijos yra sutrumpėjimai: mutacijos, kurių metu trūksta dalies baltymo, o tai gali pakeisti MECP2 ir RNR Pol II sąveiką.
Tyrėjai tikisi, kad jų išvados ne tik pakeis mūsų supratimą apie MECP2, bet ir kad gilesnis bei platesnis MECP2 įtakos smegenų vystymuisi ir funkcijai supratimas gali padėti gauti naujų įžvalgų, kurios padės žmonėms, sergantiems Retto sindromu ir susijusiais sutrikimais, įskaitant autizmą.
„Šis projektas yra puikus Janischo laboratorijos bendradarbiavimo pobūdžio pavyzdys“, – sako Flamier. „Mudu su Rudolfu turėjome specifinę problemą, susijusią su Retto sindromu, ir aš turėjau patirties su CUT&Tag technologija, kuri galėtų išspręsti šią problemą. Diskusijų metu supratome, kad galime sujungti savo pastangas ir dabar turime puikią informacijos apie MECP2 ir jo sąsajas su ligomis saugyklą.“