Medicinos ekspertas

Naujos publikacijos
Usherio sindromas
Paskutinį kartą peržiūrėta: 04.07.2025

Visas „iLive“ turinys yra peržiūrėtas medicinoje arba tikrinamas, kad būtų užtikrintas kuo didesnis faktinis tikslumas.
Mes turime griežtas įsigijimo gaires ir susiejamos tik su geros reputacijos žiniasklaidos svetainėmis, akademinių tyrimų institucijomis ir, jei įmanoma, medicininiu požiūriu peržiūrimais tyrimais. Atkreipkite dėmesį, kad skliausteliuose ([1], [2] ir tt) esantys numeriai yra paspaudžiami nuorodos į šias studijas.
Jei manote, kad bet koks mūsų turinys yra netikslus, pasenęs arba kitaip abejotinas, pasirinkite jį ir paspauskite Ctrl + Enter.
Ašerio sindromas yra paveldima liga, pasireiškianti visišku kurtumu nuo gimimo, taip pat progresuojančiu aklumu su amžiumi. Regėjimo praradimas susijęs su pigmentiniu retinitu – tinklainės pigmentinės degeneracijos procesu. Daugelis žmonių, sergančių Ašerio sindromu, taip pat turi rimtų pusiausvyros problemų.
Epidemiologija
Tyrimo dėka pavyko nustatyti, kad Usherio sindromas paveikia apie 8 % tirtų kurčiųjų-nebylių vaikų (tyrimai buvo atlikti specialiose įstaigose kurtiesiems-nebyliams). Pigmentinis retinitas pastebėtas 6–10 % pacientų, kenčiančių nuo įgimto kurtumo, o tai savo ruožtu stebima apie 30 % žmonių, sergančių pigmentine tinklainės liga.
Manoma, kad ši liga pasireiškia maždaug 3–10 žmonių iš 100 tūkstančių visame pasaulyje. Ji vienodai gali pasireikšti tiek moterims, tiek vyrams. Šiuo sindromu serga apie 5–6 % pasaulio gyventojų. Apie 10 % visų vaikystės gilaus kurtumo atvejų atsiranda dėl I, taip pat ir II tipo Usherio sindromo.
Jungtinėse Amerikos Valstijose dažniausiai pasitaiko 1 ir 2 tipai. Kartu jie sudaro maždaug 90–95 procentus visų Usherio sindromo atvejų vaikams.
Priežastys Usherio sindromas
Ašerio sindromo I, II ir III tipai yra autosominiu recesyviniu būdu paveldimi, o IV tipas laikomas X chromosomos sutrikimu. Šio sindromo sukelto aklumo ir kurtumo priežastys dar nėra pakankamai ištirtos. Manoma, kad sergantieji šia liga yra padidėję jautrumu komponentams, kurie gali pažeisti DNR struktūrą. Be to, ši liga gali būti susijusi su imuninės sistemos sutrikimais, tačiau šiuo atveju nėra tikslaus šio proceso vaizdo.
1989 m. pirmą kartą II tipo liga sergantiems pacientams buvo nustatyti chromosomų sutrikimai, kurie ateityje gali padėti išskirti sindromą sukeliančius genus. Taip pat gali būti įmanoma identifikuoti šiuos genus nešiotojams ir sukurti specialius prenatalinius genetinius testus.
 [ 8 ]
[ 8 ]
Rizikos veiksniai
Sindromas paveldimas, kai serga abu tėvai, t. y. jis paveldimas recesyviniu būdu. Vaikas taip pat gali paveldėti ligą, jei jo tėvai yra geno nešiotojai. Jei abu būsimi tėvai turi šį geną, tikimybė pagimdyti kūdikį su šiuo sindromu yra 1 iš 4. Asmuo, turintis tik vieną sindromo geną, laikomas nešiotoju, tačiau nejaučia sutrikimo simptomų. Šiais laikais dar neįmanoma nustatyti, ar žmogus turi šios ligos geną.
Jei vaikas gimsta tėvams, iš kurių vienas neturi tokio geno, tikimybė, kad jis paveldės sindromą, yra labai maža, tačiau jis tikrai bus nešiotojas.
Simptomai Usherio sindromas
Ašerio sindromo simptomai yra klausos praradimas ir nenormalus pigmentinių ląstelių kaupimasis akių struktūrose. Pacientui išsivysto tinklainės degeneracija, dėl kurios pablogėja regėjimas, o sunkiausiais atvejais – regėjimo praradimas.
Sensorineuralinis klausos praradimas gali būti lengvas arba visiškas ir paprastai neprogresuoja nuo gimimo. Tačiau tinklainės pigmento liga gali pradėti vystytis vaikystėje ar vėliau. Tyrimų rezultatai parodė, kad centrinis regėjimo aštrumas gali būti išlaikytas daugelį metų, net ir pablogėjus periferiniam regėjimui (būklė, vadinama „tuneliniu regėjimu“).
Tai yra pagrindinės ligos apraiškos, kurias kartais gali papildyti kiti sutrikimai, tokie kaip psichozė ir kiti psichikos sutrikimai, vidinės ausies problemos ir (arba) katarakta.
Formos
Tyrimo metu buvo nustatyti 3 šios ligos tipai, taip pat 4-oji forma, kuri yra gana reta.
I tipo ligai būdingas įgimtas visiškas kurtumas, taip pat pusiausvyros sutrikimas. Dažnai tokie vaikai pradeda vaikščioti tik sulaukę 1,5 metų. Regėjimo blogėjimas paprastai prasideda nuo 10 metų, o galutinis naktinio aklumo išsivystymas prasideda nuo 20 metų. Vaikams, sergantiems šia liga, gali progresuoti periferinio regėjimo blogėjimas.
Sergant II tipo liga, stebimas vidutinio sunkumo arba įgimtas kurtumas. Šiuo atveju dalinio kurtumo pablogėjimas dažnai nebepasireiškia. Pigmentinis retinitas pradeda vystytis maždaug paauglystės pabaigoje arba po 20 metų. Naktinis aklumas paprastai prasideda 29–31 metų amžiaus. Regėjimo aštrumo sutrikimas sergant II tipo patologija paprastai progresuoja šiek tiek lėčiau nei sergant I tipu.
III tipo ligai būdingas progresuojantis klausos praradimas, paprastai prasidedantis brendimo metu, taip pat laipsniškas tuo pačiu laikotarpiu (šiek tiek vėliau nei klausos praradimas) išsivystantis pigmentinis retinitas, kuris gali tapti progresuojančio aklumo vystymosi veiksniu.
IV tipo patologijos apraiškos daugiausia pasireiškia vyrams. Šiuo atveju taip pat stebimi progresuojantys sutrikimai ir klausos bei regėjimo praradimas. Ši forma yra labai reta ir paprastai turi X chromosominį pobūdį.
Diagnostika Usherio sindromas
Usherio sindromo diagnozė nustatoma remiantis paciento pastebėtu staigaus kurtumo ir progresuojančio regėjimo praradimo deriniu.
Testai
Mutacijai nustatyti gali būti paskirtas specialus genetinis tyrimas.
Nustatyti vienuolika genetinių lokusų, galinčių sukelti Usherio sindromą, ir nustatyti devyni genai, kurie neabejotinai yra šio sutrikimo priežastis:
- 1 tipas: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- 2 tipas: ush2a, VLGR1, WHRN.
- Usherio sindromas 3 tipas: USH3A.
NIDCD mokslininkai kartu su kolegomis iš Niujorko ir Izraelio universitetų nustatė Pcdh15 geno mutaciją, vadinamą R245X, kuri lemia didelę 1 tipo Usherio sindromo dalį žydų populiacijoje.
Norėdami sužinoti apie laboratorijas, atliekančias klinikinius tyrimus, apsilankykite https://www.genetests.org ir laboratorijų kataloge ieškokite „Usherio sindromas“.
Norėdami sužinoti apie esamus klinikinius tyrimus, kuriuose atliekami genetiniai Usherio sindromo tyrimai, apsilankykite https://www.clinicaltrials.gov ir ieškokite „Usherio sindromas“ arba „Usherio sindromo genetinis tyrimas“.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentinė diagnostika
Yra keli instrumentinės diagnostikos metodai:
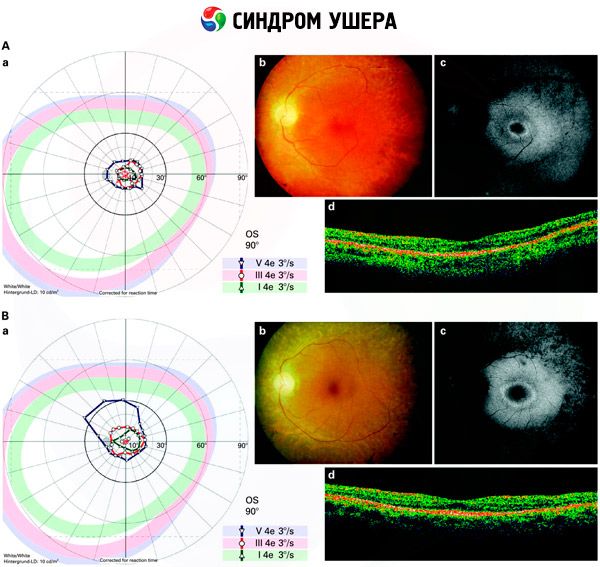
- Akies dugno tyrimas, siekiant nustatyti pigmentinių dėmių buvimą tinklainėje, taip pat tinklainės kraujagyslių susiaurėjimą;
- Elektroretinograma, leidžianti aptikti pradinius degeneracinius tinklainės nukrypimus. Rodo elektroradiografinių takų išnykimą;
- Elektronistagmograma (ENG) matuoja nevalingus akių judesius, kurie gali rodyti disbalanso buvimą.
- Audiometrija, naudojama kurtumo buvimui ir jo sunkumui nustatyti.
Diferencialinė diagnostika
Usherio sindromą reikia atskirti nuo kai kurių panašių sutrikimų.
Hallgreno sindromas, kuriam būdingas įgimtas klausos praradimas ir progresuojantis regėjimo praradimas (taip pat išsivysto katarakta ir nistagmas). Papildomi simptomai yra ataksija, psichomotoriniai sutrikimai, psichozė ir protinis atsilikimas.
Alstromo sindromas – paveldima liga, kurios metu degeneruoja tinklainė ir prarandamas centrinis regėjimas. Šis sindromas susijęs su vaikų nutukimu. Tuo pačiu metu po 10 metų pradeda vystytis cukrinis diabetas ir klausos praradimas.
Raudonukė nėščiai moteriai pirmąjį trimestrą gali sukelti įvairių vaiko vystymosi sutrikimų. Tarp tokių sutrikimų pasekmių yra klausos praradimas, taip pat (arba) regėjimo problemos, be to, įvairūs vystymosi defektai.
Su kuo susisiekti?
Gydymas Usherio sindromas
Šiuo metu nėra Usherio sindromo išgydymo. Todėl šiuo atveju terapija daugiausia susideda iš regėjimo praradimo proceso sulėtinimo ir klausos praradimo kompensavimo. Galimi gydymo metodai:
- Vartojant vitaminą A (kai kurie oftalmologai mano, kad didelės vitamino A palmitato dozės gali sulėtinti, bet ne sustabdyti pigmentinio retinito progresavimą);
- Specialių elektroninių prietaisų implantavimas į paciento ausis (klausos aparatai, kochleariniai implantai).
Oftalmologai rekomenduoja daugumai suaugusiųjų, sergančių dažnomis pigmentinio retinito formomis, kasdien vartoti 15 000 TV (tarptautinių vienetų) vitamino A palmitato prižiūrint gydytojui. Kadangi tyrime nebuvo įtraukti žmonės, sergantys 1 tipo Usherio sindromu, šiai pacientų grupei nerekomenduojamos didelės vitamino A dozės. Žmonės, svarstantys apie vitamino A vartojimą, turėtų aptarti šį gydymo būdą su savo gydytoju. Kitos rekomendacijos dėl šio gydymo būdo:
- Pakeiskite savo mitybą, įtraukdami maisto produktus, kuriuose gausu vitamino A.
- Moterys, planuojančios pastoti, turėtų nutraukti didelių vitamino A dozių vartojimą likus trims mėnesiams iki planuojamo pastojimo dėl padidėjusios apsigimimų rizikos.
- Nėščios moterys turėtų nutraukti didelių vitamino A dozių vartojimą dėl padidėjusios apsigimimų rizikos.
Taip pat svarbu tokį vaiką pritaikyti socialiniame gyvenime. Tam reikalinga specialiojo ugdymo mokytojų ir psichologų pagalba. Tuo atveju, jei pacientas pradėjo progresuoti regėjimo praradimu, jį reikėtų mokyti vartoti gestų kalbą.
Prognozė
Ašerio sindromo prognozė nepalanki. Daugumai pacientų, sergančių šia liga, regėjimo laukas ir jo aštrumas pradeda blogėti per 20–30 metų. Kai kuriais atvejais atsiranda visiškas abipusis regėjimo praradimas. Klausos praradimas, kuris visada lydimas nebylumo, labai greitai išsivysto į visišką abipusį klausos praradimą.