Medicinos ekspertas

Naujos publikacijos
Angelmano sindromas vaikams ir suaugusiesiems
Paskutinį kartą peržiūrėta: 04.07.2025

Visas „iLive“ turinys yra peržiūrėtas medicinoje arba tikrinamas, kad būtų užtikrintas kuo didesnis faktinis tikslumas.
Mes turime griežtas įsigijimo gaires ir susiejamos tik su geros reputacijos žiniasklaidos svetainėmis, akademinių tyrimų institucijomis ir, jei įmanoma, medicininiu požiūriu peržiūrimais tyrimais. Atkreipkite dėmesį, kad skliausteliuose ([1], [2] ir tt) esantys numeriai yra paspaudžiami nuorodos į šias studijas.
Jei manote, kad bet koks mūsų turinys yra netikslus, pasenęs arba kitaip abejotinas, pasirinkite jį ir paspauskite Ctrl + Enter.

Yra nemažai ligų, kurioms būdingi posakiai „rūpinkis savimi ir nesusirgsi“ skamba mažų mažiausiai juokingai. Tai patologijos, kai kai kurie psichiniai ir fiziniai sutrikimai yra būdingi vaiko organizmui dar prieš gimimą, tačiau tėvai dėl to nėra kalti. Tokias ligas sukelia chromosomų rinkinių mutacijos arba sutrikimai ir jos vadinamos chromosominėmis arba genetinėmis. Angelmano sindromas, Dauno sindromas, Patau sindromas, Edvardso sindromas, Turnerio sindromas, Praderio-Vilio sindromas – tai tik dalis genetinių ligų iš gana neblogo sąrašo.
Laimingo žmogaus sindromas
Šį kartą kalbėsime apie patologiją, pavadintą anglų pediatro Harry Angelmano vardu. Jis pirmą kartą iškėlė šios problemos klausimą 1965 m., dieną prieš tai savo praktikoje susidūręs su trimis neįprastais vaikais, kuriuos vienijo bendri savotiški simptomai. Gydytojas šiuos vaikus pavadino lėlių vaikais ir parašė apie juos straipsnį, kuris iš pradžių vadinosi „Vaikai-marionetės“. Pats straipsnis ir jo pavadinimas buvo parašyti remiantis paveikslu, matytu viename iš Veronos muziejų. Paveiksle buvo pavaizduotas besijuokiantis berniukas, o jis buvo pavadintas „Lėlininku“. Paveiksle pavaizduoto vaiko susiejimas su trimis vaikais, su kuriais Angelmanas kadaise susidūrė savo praktikoje, paskatino pediatrą sujungti vaikus į vieną grupę dėl jų ligos.
Nieko stebėtino tame, kad straipsnyje minimų vaikų nepastebėjo kiti gydytojai. Juk iš pirmo žvilgsnio atrodė, kad jie serga visiškai skirtingomis ligomis, toks skirtingas buvo bendras ligos klinikinis vaizdas 3 skirtingais atvejais. Galbūt „nauja“ chromosomų patologija būtų sudominusi ir kitus mokslininkus, tačiau tuo metu genetika dar nebuvo pakankamai išsivysčiusi, kad patvirtintų anglų gydytojo hipotezę. Todėl, po tam tikro susidomėjimo ja, straipsnis ilgam buvo numestas į galinę lentyną.
Kitas Angelmano sindromo paminėjimas, kuriuo dabar vadinamas anglų pediatro G. Angelmano straipsnis, datuojamas XX a. devintojo dešimtmečio pradžia. Ir tik 1987 m. pavyko rasti priežastį, kodėl nedidelė dalis vaikų gimsta su tokiais nukrypimais, kad iš išorės atrodo, jog jie nuolat šypsosi ir yra laimingi. Tiesą sakant, tai visai netiesa, o šypsena tėra grimasa, už kurios slepiasi nelaiminga žmogaus siela ir tėvų skausmas.
Epidemiologija
Remiantis statistika, vaiko chromosomų mutacija gali išsivystyti tiek esant panašioms tėvų mutacijoms, tiek nesant tokių. Nėra aiškaus paveldimo Angelmano sindromo (AS) pobūdžio, tačiau patologijos išsivystymo tikimybė tėvams, turintiems chromosomų mutacijų, yra gana didelė.
Taip pat įdomu, kad jei šeimoje jau yra vaikas, sergantis AS, yra vieno procento tikimybė susilaukti antro vaiko su tuo pačiu sutrikimu, net jei tėvai yra sveiki.
Vis dar nėra tikslios statistikos apie Angelmano sindromo pacientų skaičių. Galbūt priežastis yra simptomų įvairovė, kurie gali pasireikšti tam tikra sudėtimi arba ilgą laiką visai nepasireikšti. Manoma, kad ligos paplitimas yra: 1 vaikas 20 000 naujagimių. Tačiau šis skaičius yra labai apytikslis.
Priežastys Angelmano sindromas
Angelmano sindromas yra medicininis chromosomų patologijos pavadinimas, tačiau jis toli gražu ne vienintelis. Žmonės šią ligą vadina lėlių vaikų sindromu, laimingų lėlių sindromu, Petruškos sindromu ir besijuokiančios lėlės sindromu. Žmonės sugalvoja visokių pavadinimų (kartais net įžeidžiančių pačius pacientus ir jų tėvus), tačiau liga yra liga, kad ir kokia juokinga ji atrodytų ir kokios būtų priežastys.
Angelmano sindromo, kaip ir daugelio kitų genetinių patologijų, išsivystymo priežastys visais atvejais yra vienos iš chromosomų arba viso chromosomų rinkinio struktūros sutrikimai. Tačiau mūsų atveju visa problema slypi 15-oje chromosomoje, perduodamoje iš motinos. Tai yra, tėvo chromosoma šiuo atveju neturi nukrypimų, tačiau moteriškoji patiria tam tikras mutacijas.
Pagal chromosomų anomalijos tipą Angelmano sindromas klasifikuojamas kaip chromosomų mutacija. Tokios mutacijos laikomos:
- Delecija (chromosomos dalies, kurioje yra tam tikras genų rinkinys, nebuvimas; jei trūksta vieno iš genų, kalbame apie mikrodeleciją), kuri atsiranda po dviejų lūžių ir vieno susijungimo, kai prarandama pradinės chromosomos dalis.
- Dublikacija (papildomos chromosomos dalies, kuri yra esamos kopija, buvimas), kuri daugeliu atvejų veda prie žmogaus mirties, rečiau – nevaisingumo.
- Inversija (vienos iš chromosomos dalių apvertimas 180 laipsnių kampu, t. y. priešinga kryptimi, ir tada joje esantys genai išsidėstę priešinga tvarka), kai nutrūkę chromosomos galai susijungia kita nei pradinė tvarka.
- Įterpimas (jei dalis genetinės medžiagos chromosomoje yra ne savo vietoje),
- translokacija (jei tam tikra chromosomos dalis yra prijungta prie kitos chromosomos; tokia mutacija gali būti abipusė neprarandant sekcijų).
Gavęs mutavusią chromosomą iš nieko neįtariančios motinos, vaikas pasmerktas gimti su anomalijomis. Dažniausia Angelmano sindromo priežastis vis dar laikoma motinos 15-osios chromosomos ištrynimu, kai trūksta mažos dalies. Retesnės „besijuokiančios lėlės“ sindromo mutacijos laikomos:
- perkėlimas,
- vienpusė disomija (jei vaikas gavo chromosomų porą iš tėvo, motinos chromosomos nėra),
- DNR genų mutacija, kurie yra ir pagrindinė statybinė (genetinė) medžiaga, ir instrukcijos, kaip ją teisingai naudoti (ypač ube3a geno mutacija motinos chromosomoje).
Vienos iš šių mutacijų buvimas tėvų organizme yra Angelmano sindromo išsivystymo vaikams rizikos veiksnys. Tačiau ne tik chromosomų mutacijos, bet ir genominės (kurios yra susijusios su kiekybiniais chromosomų rinkinių pokyčiais ir yra dažnesnės nei chromosominės) gali išprovokuoti ligos vystymąsi vaikui. Dažnos genominės mutacijos yra chromosomų trisomija (jei žmogaus chromosomų rinkinyje yra daugiau nei 46 chromosomos).
Kad vaikui pasireikštų patologija, nebūtina, kad tėvai turėtų chromosomų anomalijų. Ir vis dėlto yra tam tikras procentas pacientų, kurių liga yra paveldima.
Pathogenesis
Pasinerkime šiek tiek giliau į biologiją, tiksliau, genetiką. Kiekvieno žmogaus organizmo genetinė informacija yra 23 chromosomų porose. Viena chromosoma iš poros vaikui perduodama iš tėvo, kita – iš motinos. Visos chromosomų poros skiriasi forma ir dydžiu bei neša tam tikrą informaciją. Taigi, 23-ioji chromosomų pora (X ir Y chromosomos) yra atsakinga už kūdikio lytinių požymių formavimąsi (XX – mergaitė, XY – berniukas, o Y chromosomą vaikas gali gauti tik iš tėvo).
Idealiu atveju vaikas iš tėvų gauna 46 chromosomas, kurios formuoja jo genetines savybes, nulemiančias jį kaip individą. Didesnis chromosomų skaičius vadinamas trisomija ir laikomas nukrypimu nuo normos. Pavyzdžiui, 47 chromosomos buvimas chromosomų rinkinyje (kariotipas, lemiantis rūšį ir individualias savybes) sukelia Dauno sindromo atsiradimą.
Jei chromosomos nudažomos specialiais dažais, mikroskopu išilgai kiekvienos iš jų galima pamatyti skirtingų atspalvių juosteles. Kiekvienos juostelės viduje yra didžiulis skaičius genų. Visos šios juostelės yra mokslininkų sunumeruotos ir turi fiksuotą vietą. Vienos juostelės nebuvimas laikomas nukrypimu nuo normos. Sergant Angelmano sindromu, labai dažnai galima pastebėti motinos chromosomos segmentų nebuvimą intervale q11-q13, esančiame ilgojoje rankoje, kurioje DNR bazių skaičius yra tik apie 4 milijonus.
Pagrindiniu chromosomos komponentu laikoma neįtikėtinai ilga DNR molekulė, turinti tūkstančius genų ir dešimtis bei šimtus milijonų azoto bazių. Taigi, 15 chromosomoje, atsakingoje už Angelmano sindromo ir kelių kitų išsivystymą, yra 1200 genų ir apie 100 milijonų bazių. Bet kokie DNR molekulės struktūros sutrikimai neabejotinai paveiks būsimo vaiko išvaizdą ir vystymąsi.
Genetinė informacija, esanti genuose, paverčiama baltymu arba RNR. Šis procesas vadinamas genų raiška. Tokiu būdu iš tėvų gauta genetinė informacija įgauna ir formą, ir turinį, kuris įkūnijamas jų unikaliame moteriškos arba vyriškos lyties įpėdinyje.
Yra nemažai patologijų, turinčių neklasikinį paveldėjimo tipą, įskaitant Angelmano sindromą, kai iš tėvų gauti genai kaip suporuotų chromosomų dalis turi unikalų tėvų įspaudą ir pasireiškia skirtingais būdais.
Taigi, Angelmano sindromas yra ryškus genominio imprintingo pavyzdys, kai genų raiška vaiko organizme tiesiogiai priklauso nuo to, iš kurio tėvo buvo gauti aleliai (skirtingos vieno geno formos, gautos iš tėvo ir motinos, esančios identiškuose suporuotų chromosomų skyriuose). Tai yra, tik motinos chromosomos anomalijos lemia sindromo vystymąsi, o tėvo chromosomos mutacijos ir struktūriniai sutrikimai sukelia visiškai skirtingas patologijas.
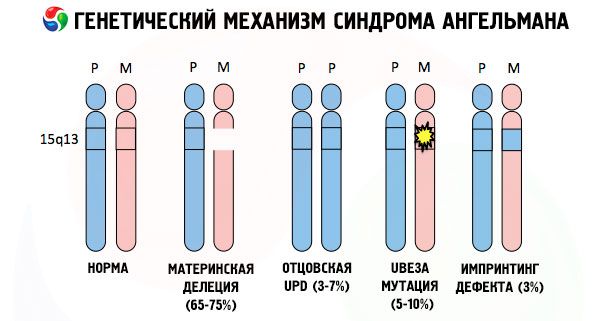
Sergant šia patologija, motinos chromosomoje trūksta tam tikrų genų arba sumažėja atskirų genų aktyvumas (didžiąja dauguma atvejų tai ube3a genas, dalyvaujantis ubikvitino, baltymo, reguliuojančio kitų baltymų skaidymą, metabolizme). Dėl to vaikui diagnozuojami protinės raidos sutrikimai ir fiziniai deformacijos.
Simptomai Angelmano sindromas
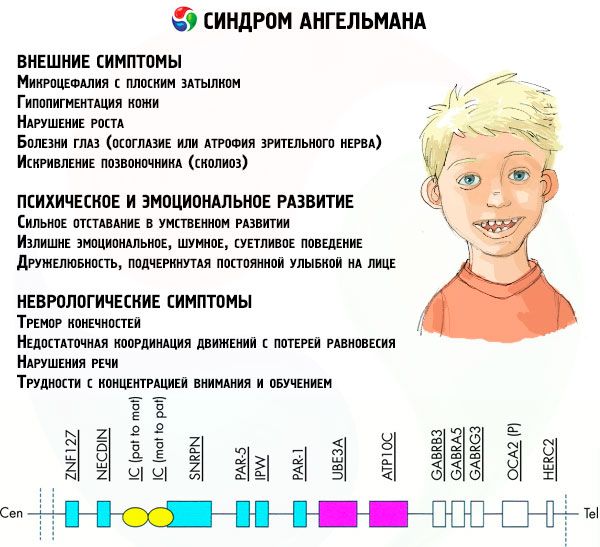
Angelmano sindromo simptomai veikia įvairius vaiko gyvenimo ir vystymosi aspektus: fizinį, neurologinį, psichinį. Remiantis tuo, galima nustatyti 3 simptomų grupes, kurios rodo šios patologijos vystymąsi.
- Išoriniai arba fiziniai simptomai:
- neproporcingai maža galva, palyginti su kūnu ir galūnėmis, kurios yra normalaus dydžio,
- per plati burna,
- beveik visada veide yra šypsena (su atvira burna),
- reti dantys,
- siaura viršutinė lūpa,
- dažnai išsikišęs platus liežuvis,
- išsikišęs apatinis žandikaulis,
- smailus smakras,
- labai šviesi oda, dažnai plaukai (albinizmas, susijęs su tuo, kad organizmas negamina pigmento melanino),
- tamsios dėmės ant šviesios odos (hipopigmentacija dėl nepakankamos melanino gamybos)
- fiziniai ar išoriniai simptomai: akių ligos, tokios kaip žvairumas ar regos nervo atrofija,
- stuburo iškrypimas (skoliozė),
- sustingusios kojos (vaikščiodamas žmogus nesulenkia kojų per kelius dėl mažo sąnarių judrumo, todėl palyginama su lėlės eisena).
- Su psichine ir emocine raida susiję simptomai:
- sunkus protinis atsilikimas,
- pernelyg emocingas, triukšmingas, irzlus elgesys,
- dažnas rankų plojimas,
- išreikštas draugiškumas, pabrėžiamas nuolatine šypsena veide,
- dažnas juokas be jokios priežasties.
- Neurologiniai simptomai:
- galūnių drebulys,
- nepakankamas judesių koordinavimas su pusiausvyros praradimu,
- sumažėjęs raumenų tonusas,
- įvairūs miego sutrikimai,
- dažni isteriniai priepuoliai vaikystėje,
- kalbos sutrikimai (vaikas pradeda kalbėti vėlai, turi prastus bendravimo įgūdžius ir nerišlią kalbą),
- hiperaktyvumas padidėjusio jaudrumo fone,
- sunkumų susikaupiant ir mokantis.
Tačiau tai yra apibendrintas ligos vaizdas. Iš tiesų, Angelmano sindromo klinikinis vaizdas labai priklauso nuo ligos vystymosi stadijos ir chromosomų mutacijos, sukėlusios patologiją, tipo. Tai reiškia, kad ligos simptomai skirtingiems pacientams gali labai skirtis, o tai ilgą laiką neleido atskirti patologijos nuo kitų, turinčių panašų klinikinį vaizdą.
Tarp visų simptomų skaičiaus galime išskirti tuos, kurie būdingi visiems pacientams be išimties:
- sunkus protinis atsilikimas,
- netinkamas elgesys (nepagrįstas juokas, padidėjęs jaudrumas, prasta koncentracija, euforijos būsena),
- motorinių įgūdžių neišsivystymas,
- prasta judesių koordinacija, eisenos ataksija (netolygus tempas, sūpavimas iš vienos pusės į kitą ir kt.), galūnių drebulys.
- kalbos raidos sutrikimas, kai vyrauja neverbalinės bendravimo priemonės.
Tarp simptomų, su kuriais susiduria dauguma pacientų, galima išskirti šiuos:
- galvos ir kūno disproporcija dėl uždelsto fizinio vystymosi,
- daugeliui pacientų kaukolės forma yra tokia, kad smegenų dydis išlieka mažesnis nei sveikų žmonių (mikrocefalija),
- epilepsijos priepuoliai iki 3 metų amžiaus, kurių stiprumas ir dažnis palaipsniui mažėja vyresniame amžiuje,
- EEG parametrų iškraipymas (žemo dažnio bangų svyravimai ir didelė amplitudė).
Šie simptomai yra gana dažni, tačiau 20 % pacientų, sergančių Angelmano sindromu, jų neturi.
Dar rečiau galima diagnozuoti tokias ligos apraiškas kaip:
- sunkus arba lengvas strabizmas,
- prasta liežuvio judesių kontrolė, dėl kurios pacientai dažnai be jokios priežasties iškiša liežuvį,
- sunkumai rijant ir čiulpiant, ypač mažiems vaikams,
- odos ir akių pigmentacijos sutrikimas,
- rankos pakeltos arba sulenktos einant,
- hiperrefleksija,
- miego sutrikimai, ypač vaikystėje,
- dažnas seilėtekis,
- nenumaldomas troškulys,
- pernelyg aktyvūs kramtymo judesiai,
- padidėjęs jautrumas karščiui,
- plokščia pakaušio dalis,
- išsikišęs apatinis žandikaulis,
- lygūs delnai.
Gana didelė dalis pacientų turi problemų su šlapinimusi, kurį jie prastai kontroliuoja, sutrikusią smulkiąją motoriką, dėl kurios sunku rūpintis savimi ir mokytis, bei turi antsvorio. Beveik visi pacientai brendimą patiria vėliau nei sveiki bendraamžiai.
Angelmano sindromą turintys vaikai gerai suvokia žodinę kalbą ir ją supranta, tačiau nenori dalyvauti pokalbyje, apribodami savo kalbą iki kelių dešimčių žodžių, reikalingų kasdieniame gyvenime. Tačiau suaugus tokie pacientai atrodo jaunesni už savo bendraamžius be genetinių patologijų.
Daugelis Angelmano sindromo simptomų yra nepastovūs, todėl klinikinis ligos vaizdas su amžiumi labai keičiasi. Traukuliai ir epilepsijos priepuoliai tampa retesni arba visiškai išnyksta, pacientas tampa mažiau jaudinamas, pagerėja miegas.
Komplikacijos ir pasekmės
Angelmano sindromas yra sunki, šiuo metu praktiškai nepagydoma chromosomų patologija, atimanti iš pacientų galimybę gyventi normalų gyvenimą. Koks bus vaiko, sergančio AS, gyvenimas, labai priklauso nuo chromosomų anomalijos tipo.
Daugeliu atvejų chromosomos segmento dubliavimasis nesuderinamas su gyvenimu. Ir net jei tokie pacientai nemiršta kūdikystėje ir nepasiekia brendimo, jie neturi jokių galimybių susilaukti vaikų.
Dažniausiai Angelmano sindromo atveju pasitaikančių genų dalies ištrynimas arba nebuvimas trukdo vaikui išmokti vaikščioti ir kalbėti. Tokiems vaikams pasireiškia sunkesnė protinio atsilikimo forma, dažniau pasireiškia epilepsijos priepuoliai, kurių intensyvumas yra daug didesnis nei pacientams, turintiems kitų chromosomų anomalijų.
Jei yra tik vieno geno mutacija, tinkamai atkreipiant dėmesį ir taikant tinkamą požiūrį, vaikas gali būti išmokytas savęs priežiūros, bendravimo ir sąveikos grupėje pagrindų, nors jis vis tiek atsiliks nuo savo bendraamžių vystymosi srityje.
Angelmano sindromą turintiems vaikams, kurie iš prigimties yra malonūs, svarbiausia yra tėvų meilė ir dėmesys. Tik tokiu atveju vaiko išsilavinimas duos vaisių, kad ir nedidelių. Žinoma, AS sergantys pacientai negalės mokytis įprastoje mokykloje. Jiems reikia specialių klasių, kuriose vaikai pirmiausia bus mokomi susikaupti, o vėliau palaipsniui bus suteikiamos pagrindinės mokyklos žinios.
Diagnostika Angelmano sindromas
Angelmano sindromas yra įgimta raidos patologija. Tačiau dėl tam tikrų aplinkybių dažnai neįmanoma jo diagnozuoti kūdikystėje ir ankstyvoje vaikystėje. Taip yra dėl nespecifiškumo ir silpnos simptomų raiškos kūdikiams ir vaikams iki 3 metų. Be to, ligos paplitimas mūsų šalyje nėra toks didelis, kad gydytojai išmoktų ją atpažinti tarp bendraamžių.
Angelmano sindromas kūdikiams gali pasireikšti sumažėjusiu raumenų tonusu, kuris pasireiškia maitinimosi problemomis (čiulpimo ir rijimo reflekso silpnumu), o vėliau ir sunkumais mokantis vaikščioti (tokie vaikai pradeda vaikščioti daug vėliau). Šie simptomai yra pirmieji kūdikio raidos sutrikimų požymiai, kurie gali būti susiję su chromosomų anomalija. Šią prielaidą gali patvirtinti tik genetinė analizė.
Ypatingas dėmesys skiriamas vaikams, kurių tėvai turi įvairių genominių ar chromosominių sutrikimų. Juk liga iš pradžių gali ir nepasireikšti, o laiku nustačius patologiją, pradėjus intensyviai dirbti su vaiku, galima pasiekti žymiai didesnės sėkmės mokantis, sulėtinti ligos progresavimą.
Jei tėvai turi įvairių chromosomų anomalijų, genetinė analizė atliekama dar prieš gimstant kūdikiui, nes SA yra viena iš patologijų, kurias galima aptikti embriono stadijoje.
Medžiagos genetiniams tyrimams rinkimas gali būti atliekamas dviem būdais:
- invazinis (su tam tikra rizikos dalimi, nes norint paimti amniono skysčio mėginį, būtina prasiskverbti į gimdą),
- neinvazinis (kūdikio DNR analizė iš motinos kraujo).
Tada atliekami šie tyrimai:
- Fluorescencinė in situ hibridizacija (FISH metodas) – prie tiriamos DNR prijungiamas specialiais dažais pažymėtas DNR zondas, o vėliau tiriamas mikroskopu.
- ube3a geno ir įspaustų genų mutacijų analizė,
- DNR metilinimo analizė, naudojant specialius genetikoje naudojamus metodus.
Genetiniai tyrimai suteikia gana tikslią informaciją chromosomų anomalijų atveju, o tai reiškia, kad būsimi tėvai iš anksto žino, kam ruoštis. Tačiau yra išimčių. Tam tikrai pacientų grupei, esant visiems patologiją rodantiems simptomams, tyrimų rezultatai išlieka normalūs. Tai yra, patologiją galima nustatyti tik atidžiai stebint vaiką nuo ankstyvos vaikystės: kaip jis valgo, kada pradėjo vaikščioti ir kalbėti, ar eidamas lenkia kojas ir pan.
Be FISH metodo, tarp instrumentinių Angelmano sindromo diagnostikos metodų galima išskirti tomografiją (KT arba MRT), kuri padeda nustatyti smegenų būklę ir dydį, bei elektroencefalogramą (EEG), kuri rodo, kaip veikia atskiros smegenų dalys.
Gydytojai galutinę diagnozę paprastai nustato 3–7 metų amžiaus, kai pacientas jau turi daugumą simptomų ir matoma ligos vystymosi dinamika.
Kokie testai reikalingi?
Diferencialinė diagnostika
Angelmano sindromas yra genetinė patologija, kuri praktiškai neturi specifinių apraiškų. Dauguma simptomų gali vienodai rodyti tiek AS, tiek kitas genetines patologijas.
Angelmano sindromo diferencinė diagnozė atliekama su šiomis patologijomis:
- Pitt-Hopkins sindromas (pacientams būdingas protinis atsilikimas, linksmas charakteris, besišypsantys, jie turi gana didelę ir plačią burną, pastebima mikrocefalija). Skirtumas yra hiperventiliacijos ir kvėpavimo sulaikymo priepuoliai budrumo būsenoje.
- Christiansono sindromas (pacientai yra protiškai atsilikę žmonės, linksmo būdo, negalintys kalbėti, kuriems būdinga mikrocefalija, ataksija, traukuliai, nevalingi raumenų judesiai).
- Mowat-Wilson sindromas (simptomai: protinis atsilikimas, epilepsijos priepuoliai, smailus smakras, atvira burna, laiminga veido išraiška, mikrocefalija). Skiriamieji bruožai: didelis atstumas tarp akių, į vidų pakreiptos akys, apvalus nosies galiukas, atgal atlenkta ausis.
- Kabuki sindromas (būdingas lengvas arba vidutinio sunkumo protinis atsilikimas, kalbos ir motorikos sutrikimai, raumenų silpnumas, epilepsijos priepuoliai, mikrocefalija, ilgi intervalai tarp niežulių ir sutrikusi koordinacija). Jam būdingi išlenkti antakiai, išvirkščia apatinio voko šoninė dalis, plačiai išdėstytos akys, ilgi vokų įtrūkimai su ilgomis, storomis blakstienomis.
- Retto sindromas (skirtumas nuo AS moterims). Simptomai: uždelsta kalbos raida, traukuliai, mikrocefalija. Skirtumas tas, kad veide nėra laimingos išraiškos, atsiranda apnėjos ir apraksijos priepuoliai, kurie laikui bėgant progresuoja.
- Autosominis recesyvinis protinio atsilikimo sindromas 38 (simptomai: ryškus protinis atsilikimas su motorinių įgūdžių ir kalbos atsilikimu, raumenų silpnumas, maitinimosi problemos kūdikystėje, impulsyvumas). Skiriamasis bruožas – mėlyna rainelės spalva.
- MECP 2 geno duplikacijos sindromas (diferenciacija nuo SA vyrams). Simptomai: sunkus protinis atsilikimas, raumenų silpnumas nuo vaikystės, kalbos sutrikimai arba kalbos trūkumas, epilepsija. Skiriamieji požymiai: progresuojanti miopatija, nuolat pasikartojančios infekcijos.
- Kleefstros sindromas (simptomai: kalbos ir mąstymo sutrikimai, raumenų silpnumas, miego sutrikimai, dėmesio stoka, atvira burna, hiperaktyvumas, traukuliai, ataksija, pusiausvyros sutrikimai). Skiriamieji požymiai: plokščias veidas, trumpa kumpa nosis, plačiai išdėstytos akys, didelė išvirkščia apatinė lūpa, agresyvūs protrūkiai.
- Smith-Magenis sindromas (būdingas traukuliais, miego sutrikimais, intelekto ir motorikos sutrikimais). Būdingi bruožai yra platus ir plokščias veidas bei iškili kakta.
- Koolen-de-Vries sindromas (lengvas arba vidutinio sunkumo protinis atsilikimas, raumenų silpnumas, traukuliai, draugiškumas). Skiriamieji požymiai: pailgas veidas su aukšta kakta, atsikišusios ausys, pasvirusios akys, didelis sąnarių judrumas, įgimtos širdies ydos.
- Phelan-McDermid sindromas (simptomai: protinis atsilikimas, kalbos sutrikimai arba kalbos stoka). Skiriamieji bruožai: didelės rankos su išsivysčiusiais raumenimis, raumenų silpnumas nuo gimimo, silpnas prakaitavimas.
Tokios patologijos kaip adenilsukcinato trūkumas, autosominis recesyvinis protinio atsilikimo sindromas 1, 2q23.1 chromosomos dubliavimo sindromas, FOXG1, STXBP1 arba MEF2C geno haploinsufficiency sindromai ir kai kurie kiti gali „pasigirti“ panašiais į Angelmano sindromą simptomais.
Gydytojo užduotis – tiksliai diagnozuoti, atskirti Angelmano sindromą nuo panašių simptomų turinčių patologijų ir paskirti veiksmingą gydymą, atitinkantį diagnozuotą ligos stadiją.
Gydymas Angelmano sindromas
Angelmano sindromas yra viena iš tų patologijų, kurioms medicina vis dar ieško veiksmingo gydymo. Etiologinis ligos gydymas yra įvairių metodų ir priemonių kūrimo stadijoje, daugelis jų dar nebuvo išbandyti su žmonėmis. Tai reiškia, kad kol kas gydytojai turi apsiriboti simptominiu gydymu, kuris kažkaip padeda palengvinti nepavydėtiną marionetės sindromu sergančių vaikų ir suaugusiųjų, kenčiančių nuo epilepsijos priepuolių, seilėtekio, hipotenzijos ir miego sutrikimų, padėtį.
Taigi, tinkamai parinktu prieštraukuliniu vaistu galima sumažinti epilepsijos priepuolių dažnį ir stiprumą. Tačiau visas sunkumas yra tas, kad pacientų, sergančių SA, priepuoliai skiriasi nuo įprastų epilepsijos priepuolių tuo, kad jiems būdingi keli priepuolių tipai, o tai reiškia, kad būklę galima palengvinti vienu metu skiriant kelis vaistus.
Populiariausi prieštraukuliniai vaistai, vartojami Angelmano sindromui gydyti, yra: valproinė rūgštis, topiramatas, lamotriginas, levetiracetamas, klonazepamas ir jų pagrindu sukurti vaistai. Rečiau vartojami vaistai, kurių pagrindą sudaro karmazepinas, fenitoinas, fenobarbitalis, etosuksimidas, nes kai kurie iš jų gali sukelti paradoksalų poveikį, pasireiškiantį epilepsijos priepuolių sustiprėjimu ir padažnėjimu. Tai atsitinka, jei vaistas vartojamas kaip monoterapijos dalis.
Seilėtekiui gydyti dažniausiai taikomi du metodai: medikamentinis (vaistas, slopinantis seilių gamybą) ir chirurginis, kurio metu seilių latakai vėl implantuojami. Tačiau SA atveju šie metodai laikomi neveiksmingais, ir klausimas lieka atviras. Tėvai ir tokių pacientų slaugytojai turi atkreipti ypatingą dėmesį į šį klausimą, nes patys pacientai dažniausiai nekontroliuoja seilėtekio, o kai kurie tiesiog negali savimi pasirūpinti.
Kita problema – trumpa miego trukmė. Dažnai vaikai, sergantys Angelmano sindromu, miega ne ilgiau kaip 5 valandas, o tai neigiamai veikia viso organizmo veiklą. Lengvai susijaudinantys, aktyvūs vaikai, mėgstantys žaidimus ir bendravimą (net jei jie stengiasi apsiriboti neverbaliniais metodais), dienos metu pastebimai pavargsta. Kad kūnas gerai pailsėtų, jam reikia gilaus, visaverčio miego, tačiau būtent čia ir slypi problema.
Atrodytų, kad raminamieji vaistai (fenotiazinai ir netipiniai antipsichoziniai vaistai), kurie ramina nervų sistemą, turėtų pakakti, kad pagerėtų miegas jautriems pacientams. Tačiau AS atveju tokių vaistų vartojimas yra kupinas neigiamo poveikio. Todėl gydytojai vis dar renkasi švelnius migdomuosius vaistus, tokius kaip melatoninas (natūralus hormoninis vaistas, pagrįstas miego hormonu), kuris pacientams skiriamas valandą prieš miegą po 1 tabletę, ir difenhidraminas. Vartojimo dažnumą ir dozę nustato gydytojas, atsižvelgdamas į paciento būklę ir amžių.
Kartais pacientams, sergantiems Angelmano sindromu, kyla problemų su virškinimu ir išmatomis. Išmatas galite pagerinti vidurius laisvinančiais vaistais (geriausia vaistažolių).
Arba galite spręsti problemą kitaip, kaip tai darė Amerikos gydytojai, remdamiesi kai kuriais autizmo gydymo metodais, nes daugelis AS būdingų simptomų būdingi ir autizmui (impulsyvumas, nevalingi judesiai, pasikartojantys veiksmai, dėmesio deficitas, bendravimo problemos ir kt.). Pastebėta, kad hormono sekretino, kuris normalizuoja virškinimą ir išmatas, įvedimas teigiamai veikia pacientų dėmesį, o oksitocinas padeda pagerinti vaiko pažintinius gebėjimus ir atmintį, teisingą elgesį.
Tiesa, vien hormonų nepakanka, ypač kai kalbama apie vaikus. Esant Angelmano sindromui, indikuotina elgesio terapija, darbas su psichologu ir logopedu (mokyti neverbalinio bendravimo metodų ir gestų kalbos). Tokių vaikų ugdymas turėtų būti grindžiamas individualia programa, kurioje dalyvautų specialiai apmokyti mokytojai, psichologas ir tėvai. Deja, ne visur tai įmanoma, ir šeimos lieka vienos su savo problema.
Kadangi daugelis jaunų pacientų, sergančių AS, kenčia nuo sumažėjusio raumenų tonuso ir sąnarių problemų, daug dėmesio skiriama kineziterapijai. Dažniausiai gydytojai naudoja parafino aplikacijas, elektroforezę ir magnetinę terapiją.
Aktyvus toninis masažas ir specialūs gydomojo fizinio lavinimo pratimai padės sergančiam vaikui po kurio laiko atsistoti ant kojų ir užtikrintai vaikščioti. Šiuo atžvilgiu ypač naudinga vandens gimnastika, kuri rekomenduojama esant SA vėsiame vandenyje. Ji padidina raumenų tonusą ir moko vaiką valdyti savo kūną bei koordinuoti judesius.
Antikonvulsantinis gydymas
Pavojingiausias Angelmano sindromo simptomas yra traukuliai, panašūs į epilepsijos. Šis simptomas pasireiškia 80% pacientų, todėl visiems jiems reikia skirti veiksmingą gydymą nuo traukulių.
Epilepsijos priepuolių gydymas atliekamas vitaminais ir prieštraukuliniais vaistais. Angelmano sindromo, lydimo konvulsinio sindromo, atveju bus naudingi B grupės vitaminai, taip pat vitaminai C, D ir E. Tačiau savarankiškai skirti vitaminų terapiją šiuo atveju yra labai pavojinga, nes nekontroliuojamas vitaminų vartojimas gali sumažinti priešepilepsinių vaistų veiksmingumą ir išprovokuoti naujus, sunkesnius ir ilgesnius priepuolius.
Prieštraukulinių vaistų parinkimą ir jų veiksmingą dozę taip pat turėtų atlikti specialistas. Jis taip pat nusprendžia, ar pakaks vieno vaisto, ar pacientui teks vartoti 2 ar daugiau vaistų ilgą laiką.
Daugumai pacientų gydytojai skiria valproinės rūgšties preparatus (Valproinę rūgštį, Depakiną, Konvulexą, Valpariną ir kt.), kurie apsaugo nuo traukulių ir pagerina pacientų nuotaiką bei psichinę būklę.
Valproinė rūgštis tiekiama tablečių, sirupo ir injekcinių tirpalų pavidalu. Populiariausias vaistas yra pailginto atpalaidavimo vaistas „Depakine“ tabletėmis ir kaip tirpalas į veną. Vaisto dozę nustato gydytojas individualiai, atsižvelgdamas į paciento svorį, amžių ir būklę.
Vaistas vartojamas valgio metu 2–3 kartus per dieną. Vidutinė paros dozė yra 20–30 mg 1 kilogramui paciento svorio, didžiausia – 50 mg/kg per parą.
Kontraindikacijos vartoti. Nevartoti esant kepenų ir kasos funkcijos sutrikimui, hemoraginei diatezei, hepatitui, porfirijai ir padidėjusiam jautrumui vaistui.
Šalutinis poveikis yra rankų drebulys, virškinimo ir išmatų sutrikimai bei kūno svorio pokyčiai.
„Topiramatas“ taip pat yra pasirinktas vaistas nuo SA. Jis tiekiamas tablečių pavidalu ir vartojamas tiek kaip monoterapijos dalis, tiek kartu su kitais vaistais.
Vartojimo būdas ir dozavimas. Tabletes gerti per burną, neatsižvelgiant į valgį. Pradinė paros dozė suaugusiesiems yra 25–50 mg, vaikams – 0,5–1 mg/kg. Kiekvieną savaitę dozė didinama gydytojo nurodymu.
Vaisto negalima vartoti nėštumo ir žindymo laikotarpiu, taip pat esant padidėjusiam jautrumui jo sudedamosioms dalims. Vaistas turi daug įvairių šalutinių poveikių.
Vaistai, kuriuos gydytojas gali skirti Angelmano sindromui gydyti: klomazepamas, rivotrilis, lamotriginas, seizaras, lamictalas, levetiracetamas, keppra, epiterra ir kt.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Tradicinė medicina ir homeopatija
Tradicinė medicina, kaip ir homeopatiniai preparatai, žinoma, yra gana saugi, tačiau tokio gydymo veiksmingumas Angelmano sindromui gali būti laikomas prieštaringu.
Nors liaudiškas gydymas vis tiek gali padėti kai kuriais atvejais. Kalbame apie epilepsijos priepuolių sustabdymą. Šiuo atžvilgiu vaistažolių gydymas gali būti gana veiksmingas.
Gerą poveikį suteikia vaistinis preparatas, kurio pagrindą sudaro bijūnas, saldymedis ir ančiuviai (komponentai imami lygiomis dalimis). Žoleles reikia sumalti į miltus. Praėjus 2 savaitėms nuo vartojimo pradžios, galima pastebėti reikšmingą priepuolių dažnio sumažėjimą.
Levandų nuoviras (1 arbatinis šaukštelis stiklinei verdančio vandens) taip pat naudingas nuo mėšlungio. Mišinys virinamas 5 minutes ir užpilamas pusvalandį. Vaistas geriamas naktį 14 dienų.
Vandeninė (arba alkoholinė) motinėlės užpilas laikomas veiksmingu epilepsijos priepuoliams gydyti.
Iš homeopatinių preparatų, skirtų Angelmano sindromo traukuliams išvengti, galite naudoti vaistus, kurių pagrindą sudaro ramunėlės ir sukatžolės, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Tačiau reikia nepamiršti, kad tik homeopatinis gydytojas gali skirti veiksmingas ir saugias vaistų dozes kiekvienu konkrečiu atveju.
Prevencija
Kaip skaitytojas tikriausiai jau suprato, medicina kol kas negali užkirsti kelio genų mutacijoms ir kitoms chromosomų anomalijoms, taip pat ištaisyti situacijos. Tai gali nutikti bet kam, nes vaikai, sergantys Angelmano sindromu, gimsta sveikiems tėvams, o genetika, kuri šiuo metu yra viena mažiausiai ištirtų medicinos sričių, to dar negali paaiškinti.
Vienintelis dalykas, kurį galima padaryti, tai atsakingai žiūrėti į nėštumo planavimą, laiku užsiregistruoti ir atlikti tyrimus. Tačiau vėlgi, tokia priemonė bus labiau edukacinė nei prevencinė, kaip ir bet kokie tyrimai. Tačiau jauni tėvai iš anksto žinos, kam ruoštis, ir teigiamo atsakymo atveju nuspręs, ar gali prisiimti tokią atsakomybę kaip sergančio vaiko auginimas.
Prognozė
Angelmano sindromo prognozė priklauso nuo chromosomų anomalijos pobūdžio ir jos nustatymo savalaikiškumo. Sunkiausiai nukenčia tie vaikai, kurių 15 chromosomoje yra genų „spragų“ (ištrynimas). Tikimybė, kad tokie pacientai vaikščios ir kalbės, yra itin maža. Kitus atvejus galima ištaisyti rūpestingai žiūrint į vaiką ir parodant meilę jam.
Deja, tokie pacientai negalės tapti visaverčiais visuomenės nariais, nepaisant to, kad jie toli gražu nėra kvaili, supranta kalbą ir jos reikšmę. Tačiau bendravimo problemų jie turės visą likusį gyvenimą. Pacientus galima mokyti gestų kalbos nuo vaikystės, tačiau negalima jų versti bendrauti žodžiais. „Kalbančių“ pacientų žodynas apsiriboja iki minimumo žodžių, vartojamų kasdieniame gyvenime (5–15 žodžių).
Kalbant apie Angelmano sindromo pacientų gyvenimo trukmę ir bendrą sveikatos būklę, čia pateikti skaičiai svyruoja apie vidutines vertes. Suaugusiesiems dažniausiai pasireiškia tokios sveikatos problemos kaip skoliozė ir nutukimas, kurios, tinkamai gydant, nekelia pavojaus gyvybei.