Medicinos ekspertas

Naujos publikacijos
Charcot-Marie-Tooth liga.
Paskutinį kartą peržiūrėta: 12.07.2025

Visas „iLive“ turinys yra peržiūrėtas medicinoje arba tikrinamas, kad būtų užtikrintas kuo didesnis faktinis tikslumas.
Mes turime griežtas įsigijimo gaires ir susiejamos tik su geros reputacijos žiniasklaidos svetainėmis, akademinių tyrimų institucijomis ir, jei įmanoma, medicininiu požiūriu peržiūrimais tyrimais. Atkreipkite dėmesį, kad skliausteliuose ([1], [2] ir tt) esantys numeriai yra paspaudžiami nuorodos į šias studijas.
Jei manote, kad bet koks mūsų turinys yra netikslus, pasenęs arba kitaip abejotinas, pasirinkite jį ir paspauskite Ctrl + Enter.
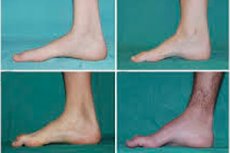
Šeivikaulio raumenų atrofija, Charcot-Marie-Tooth sindromas arba liga yra grupė lėtinių paveldimų ligų, pažeidžiančių periferinius nervus.
Pagal TLK-10, nervų sistemos ligų skyriuje šios ligos kodas yra G60.0 (paveldima motorinė ir sensorinė neuropatija). Ji taip pat įtraukta į našlaičių ligų sąrašą.
Epidemiologija
Remiantis klinikine statistika, visų tipų Charcot-Marie-Tooth ligos paplitimas 100 tūkst. gyventojų yra 19 atvejų (kitų šaltinių duomenimis, vienas atvejis 2,5–10 tūkst. gyventojų).
CMT 1 tipas sudaro maždaug du trečdalius atvejų (vienas atvejis 5 000–7 000 gyventojų), ir beveik 70 % jų yra susiję su PMP22 geno dubliavimu. Šios rūšies liga serga daugiau nei 1,2 milijono žmonių visame pasaulyje.
CMT 4 tipo paplitimas yra maždaug 1–5 atvejai 10 000 vaikų. [ 1 ]
Priežastys Šarko-Marijos-Tooto liga
Pagal polineuropatinių sindromų klasifikaciją, šeivikaulių (šeivikaulių) raumenų atrofija, Charcot-Marie-Tooth nervinė amiotrofija arba Charcot-Marie-Tooth liga (sutrumpintai CMT) reiškia genetiškai nulemtas motorines-sensorines polineuropatijas. [ 2 ]
Tai yra, jo atsiradimo priežastys yra genetinės mutacijos. Ir priklausomai nuo genetinių nukrypimų pobūdžio, išskiriami pagrindiniai šio sindromo tipai arba rūšys: demielinizacinis ir aksoninis. Pirmajai grupei priklauso 1 tipo Charcot-Marie-Tooth liga (CMT1), kuri atsiranda dėl PMP22 geno, koduojančio transmembraninį periferinį mielino baltymą 22, dubliavimosi 17 chromosomoje. Dėl to atsiranda segmentinė aksono apvalkalo (nervų ląstelių ataugų) demielinizacija ir sumažėja nervinio signalo laidumo greitis. Be to, mutacijos gali būti ir kai kuriuose kituose genuose.
Aksoninė forma yra 2 tipo Charcot-Marie-Tooth liga (CMT2), kuri pažeidžia pačius aksonus ir yra susijusi su patologiniais MFN2 geno pokyčiais 1p36.22 lokuse, koduojančiame membraninį baltymą mitofuziną-2, kuris yra būtinas mitochondrijų susiliejimui ir funkcinių mitochondrijų tinklų formavimuisi periferinių nervų ląstelėse. Yra daugiau nei dešimt CMT2 potipių (su mutacijomis specifiniuose genuose).
Pažymėtina, kad šiuo metu nustatyta daugiau nei šimtas genų, kurių pažeidimas, perduodamas paveldėjimo būdu, sukelia įvairius Charcot-Marie-Tooth ligos potipius. Pavyzdžiui, RAB7 geno mutacijos sukelia 2B tipo CMT; SH3TC2 geno (koduojančio vieną iš Schwann ląstelių membraninių baltymų) pakitimas sukelia 4C tipo CMT, kuris pasireiškia vaikystėje ir kuriam būdinga motorinių ir sensorinių neuronų demielinizacija (yra pusantros tuzino šios ligos 4 tipo formų).
Retas 3 tipo CMT (vadinamas Dejerine-Sottas sindromu) pradeda vystytis ankstyvoje vaikystėje ir yra sukeliamas PMP22, MPZ, EGR2 ir kitų genų mutacijų.
Kai 5 tipo CMT pasireiškia 5–12 metų amžiaus, stebima ne tik motorinė neuropatija (apatinių galūnių spazminės paraparezės forma), bet ir regos bei klausos nervų pažeidimas.
Raumenų silpnumas ir regos nervo atrofija (su regėjimo praradimu) bei pusiausvyros sutrikimai yra būdingi 6 tipo CMT. O 7 tipo Charcot-Marie-Tooth ligos atveju pasireiškia ne tik motorinė-sensorinė neuropatija, bet ir tinklainės liga – pigmentinis retinitas.
Dažniau vyrams pasireiškianti su X chromosoma susijusi KMT arba Charcot-Marie-Tooth liga su galūnių tetrapareze (abiejų rankų ir kojų judesių silpnumu) yra demielinizacinio tipo liga, manoma, kad ją sukelia GJB1 geno, esančio X chromosomos ilgojoje rankoje, mutacija. Šis genas koduoja koneksą 32 – Schwanno ląstelių ir oligodendrocitų transmembraninį baltymą, reguliuojantį nervinių signalų perdavimą [ 3 ].
Rizikos veiksniai
Pagrindinis CMT rizikos veiksnys yra ligos anamnezė šeimoje, tai yra, artimuose giminaičiuose.
Pasak genetikų, jei abu tėvai yra autosominiu recesyviniu Charcot-Marie-Tooth ligos geno nešiotojai, rizika susirgti šia liga yra 25 %. O rizika, kad vaikas bus šio geno nešiotojas (bet neturės jokių simptomų), yra maždaug 50 %.
Su X chromosoma susijusio paveldėjimo atveju (kai mutavusis genas yra moters X chromosomoje) yra 50 % rizika, kad motina perduos geną sūnui, kuris susirgs KMT. Liga gali nepasireikšti gimus mergaitei, tačiau dukters sūnūs (anūkai) gali paveldėti defektinį geną ir liga vystysis.
Pathogenesis
Bet kokio tipo Charcot-Marie-Tooth ligos patogenezę sukelia paveldima periferinių nervų anomalija: motorinė (judėjimo) ir sensorinė (jautri).
Jei CMT tipas yra demielinizuojantis, mielino apvalkalo, apsaugančio periferinių nervų aksonus, sunaikinimas arba defektas sulėtina nervinių impulsų perdavimą periferinėje nervų sistemoje – tarp smegenų, raumenų ir jutimo organų.
Esant aksoniniam ligos tipui, pažeidžiami patys aksonai, o tai neigiamai veikia nervinių signalų stiprumą, kurio nepakanka visiškam raumenų ir jutimo organų stimuliavimui.
Taip pat skaitykite:
Kaip perduodamas Charcot-Marie-Tooth sindromas? Defektyvūs genai gali būti paveldimi autosominiu dominantiniu arba autosominiu recesyviniu būdu.
Dažniausias tipas – autosominis dominantinis paveldėjimas – pasireiškia, kai yra viena mutavusio geno kopija (kurią nešioja vienas iš tėvų). Tikimybė perduoti CMT kiekvienam gimusiam vaikui yra maždaug 50 % [ 4 ].
Autosominio recesyvinio paveldėjimo atveju ligai išsivystyti reikalingos dvi defektinio geno kopijos (po vieną iš kiekvieno tėvo, kuris nerodo jokių ligos požymių).
40–50 % atvejų pasireiškia autosominė dominantinė paveldima demielinizacija, t. y. 1 tipo CMT; 12–26 % atvejų – aksoninė CMT, t. y. 2 tipo. O 10–15 % atvejų stebimas su X chromosoma susijęs paveldimumas [ 5 ].
Simptomai Šarko-Marijos-Tooto liga
Paprastai pirmieji šios ligos požymiai pradeda ryškėti vaikystėje ir paauglystėje ir palaipsniui vystosi visą gyvenimą, nors sindromas gali pasireikšti ir vėliau. Simptomų derinys yra įvairus, o ligos progresavimo greičio ir sunkumo numatyti neįmanoma.
Tipiniai pradinės stadijos simptomai yra padidėjęs bendras nuovargis; sumažėjęs pėdų, čiurnų ir blauzdų raumenų tonusas (silpnumas); refleksų stoka. Dėl to sunku judėti pėdai ir atsiranda disbazija (eisenos sutrikimas), pasireiškianti didesniu kojų pakilimu, dažnai su dažnais suklupimais ir kritimais. Mažo vaiko Charcot-Marie-Tooth ligos požymiai gali būti ryškus nerangumas ir amžiui nebūdingi vaikščiojimo sunkumai, susiję su abipusiu pėdos nusvirimu. Taip pat būdingos pėdos deformacijos: aukštas pėdos skliautas (įdubusi pėda) arba ryškios plokščiapėdystės, išlenkti (plaktuko formos) pirštai.
Jei vaikas vaikšto ant pirštų galų raumenų hipotonijos fone, neurologas gali įtarti, kad jam diagnozuota 4 tipo CMT, kai vaikai gali nebevaikščioti iki paauglystės.
Ligai progresuojant, raumenų atrofija ir silpnumas plinta į viršutines galūnes, todėl sunku atlikti smulkiąją motoriką ir įprastas rankų užduotis. Sumažėję lytėjimo pojūčiai ir gebėjimas jausti šilumą bei šaltį, taip pat pėdų ir rankų tirpimas rodo jutimo nervų aksonų pažeidimą.
Sergant 3 ir 6 tipų Charcot-Marie-Tooth liga, kuri pasireiškia vaikystėje, stebima sensorinė ataksija (judesių koordinacijos ir pusiausvyros sutrikimas), raumenų trūkčiojimas ir tremoras, veido nervo pažeidimas, regos nervo atrofija su nistagmu ir klausos praradimas.
Vėlesniuose etapuose gali pasireikšti nekontroliuojamas drebulys (tremoras) ir dažni raumenų mėšlungiai; judėjimo sutrikimai gali sukelti skausmą: raumenų, sąnarių, neuropatinį.
Komplikacijos ir pasekmės
Charcot-Marie-Tooth liga gali turėti komplikacijų ir pasekmių, tokių kaip:
- dažnesni patempimai ir lūžiai;
- kontraktūros, susijusios su periartikulinių raumenų ir sausgyslių sutrumpėjimu;
- skoliozė (stuburo iškrypimas);
- kvėpavimo sutrikimai – kai pažeidžiamos diafragmos raumenis inervuojančios nervinės skaidulos:
- gebėjimo judėti savarankiškai praradimas.
Diagnostika Šarko-Marijos-Tooto liga
Diagnozė apima klinikinį tyrimą, anamnezės surinkimą (įskaitant šeimos istoriją), neurologinį ir sisteminį ištyrimą.
Atliekami judesių amplitudės, jautrumo ir sausgyslių refleksų tyrimai. Nervų laidumą galima įvertinti naudojant instrumentinę diagnostiką – elektromiografiją arba elektroneuromiografiją. Taip pat gali prireikti ultragarso arba MRT. [ 6 ]
Genetiniai arba DNR tyrimai, skirti nustatyti dažniausiai pasitaikančias genetines mutacijas, sukeliančias CMT kraujo mėginyje, yra riboti, nes DNR tyrimai šiuo metu negalimi visų tipų ligai nustatyti. Daugiau informacijos žr. Genetiniai tyrimai.
Kai kuriais atvejais atliekama periferinio nervo (dažniausiai suralinio nervo) biopsija.
Diferencialinė diagnostika
Diferencinė diagnozė apima kitas periferines neuropatijas, Diušeno raumenų distrofiją, mielopatinius ir miasteninius sindromus, diabetinę neuropatiją, mielopatijas sergant išsėtine ir amiotrofine lateraline skleroze, Guillain-Barré sindromą, šeivikaulio nervo traumą ir jo atrofiją (įskaitant suspaudimą tarp juosmeninių stuburo diskų), smegenėlių ar talamo pažeidimą, taip pat chemoterapijos šalutinį poveikį (gydymo citostatikais, tokiais kaip vinkristinas ar paklitakselis, metu). [ 7 ]
Su kuo susisiekti?
Gydymas Šarko-Marijos-Tooto liga
Šiandien šios paveldimos ligos gydymas apima mankštos terapiją (skirtą stiprinti ir tempti raumenis); ergoterapiją (kuri padeda pacientams, kuriems silpni rankų raumenys); ir ortopedinių prietaisų naudojimą vaikščiojimui palengvinti. Prireikus skiriami skausmą malšinantys vaistai arba vaistai nuo traukulių. [ 8 ]
Esant sunkiai plokščiapėdystei, galima atlikti osteotomiją, o kulno deformacijos atveju – chirurginę korekciją – artrodezę. [ 9 ]
Šiuo metu atliekami tyrimai, siekiant ištirti tiek genetinį ligos komponentą, tiek jos gydymo metodus. Kamieninių ląstelių, tam tikrų hormonų, lecitino ar askorbo rūgšties naudojimas kol kas nedavė teigiamų rezultatų.
Tačiau naujausių tyrimų dėka netolimoje ateityje Charcot-Marie-Tooth ligos gydyme iš tiesų gali atsirasti kažkas naujo. Todėl nuo 2014 m. Prancūzijos bendrovė „Pharnext“ kuria, o nuo 2019 m. vidurio vyksta vaisto PXT3003, skirto 1 tipo CMT gydymui suaugusiesiems, klinikiniai tyrimai, kuris slopina padidėjusią PMP22 geno raišką, gerina periferinių nervų mielinizaciją ir palengvina neuromuskulinius simptomus.
Medicinos kompanijos „Sarepta Therapeutics“ (JAV) specialistai kuria genų terapiją 1 tipo Charcot-Marie-Tooth ligai gydyti. Šioje terapijoje bus naudojamas nekenksmingas adeno-asocijuotas virusas (AAV) iš Dependovirus genties su linijiniu vienos grandinės DNR genomu, kuris į organizmą perkels NTF3 geną, koduojantį neurotrofino-3 (NT-3) baltymą, būtiną Schwanno nervų ląstelių funkcionavimui.
„Helixmith“ iki 2020 m. pabaigos pradės Pietų Korėjoje sukurtos genų terapijos „Engensis“ (VM202) klinikinius tyrimus, skirtus 1 tipo CMT raumenų simptomams gydyti. [ 10 ]
Prevencija
KMT prevencija gali būti būsimų tėvų genetinis konsultavimas, ypač jei kas nors iš poros turi šios ligos šeimos istoriją. Tačiau buvo nustatyti de novo taškinių genų mutacijų atvejai, tai yra, kai ligos šeimos istorijoje nėra.
Nėštumo metu Charcot-Marie-Tooth ligos tikimybę negimusiam vaikui galima patikrinti atliekant chorioninio gaurelio biopsiją (nuo 10 iki 13 nėštumo savaičių), taip pat atliekant amniono skysčio analizę (15–18 savaičių).
Prognozė
Apskritai, skirtingų Charcot-Marie-Tooth ligos tipų prognozė priklauso nuo klinikinio sunkumo, tačiau visais atvejais liga progresuoja lėtai. Daugelis pacientų turi negalią, nors tai nesumažina gyvenimo trukmės.