Medicinos ekspertas

Naujos publikacijos
Paveldimas vaikų nefritas (Alporto sindromas)
Paskutinį kartą peržiūrėta: 05.07.2025

Visas „iLive“ turinys yra peržiūrėtas medicinoje arba tikrinamas, kad būtų užtikrintas kuo didesnis faktinis tikslumas.
Mes turime griežtas įsigijimo gaires ir susiejamos tik su geros reputacijos žiniasklaidos svetainėmis, akademinių tyrimų institucijomis ir, jei įmanoma, medicininiu požiūriu peržiūrimais tyrimais. Atkreipkite dėmesį, kad skliausteliuose ([1], [2] ir tt) esantys numeriai yra paspaudžiami nuorodos į šias studijas.
Jei manote, kad bet koks mūsų turinys yra netikslus, pasenęs arba kitaip abejotinas, pasirinkite jį ir paspauskite Ctrl + Enter.
Paveldimas nefritas (Alporto sindromas) yra genetiškai nulemta paveldima neimuninė glomerulopatija, pasireiškianti hematurija (kartais su proteinurija), progresuojančiu inkstų funkcijos silpnėjimu, išsivystant lėtiniam inkstų nepakankamumui, dažnai derinamu su sensorineuraliniu kurtumu ir regos sutrikimais.
Ligą pirmą kartą 1902 m. aprašė L. G. Guthrie, stebėjęs šeimą, kurioje hematurija buvo stebima keliose kartose. 1915 m. A. F. Hurstas aprašė uremijos išsivystymą tos pačios šeimos nariams. 1927 m. A. Alportas pirmą kartą nustatė klausos praradimą keliems giminaičiams, sergantiems hematurija. Šeštajame dešimtmetyje buvo aprašyti akių pažeidimai sergant panašia liga. 1972 m. pacientams, sergantiems paveldima hematurija, atlikus inkstų audinio morfologinį tyrimą, Hinglais ir kt. atskleidė netolygų glomerulų pamatinių membranų išsiplėtimą ir stratifikaciją. 1985 m. buvo nustatytas paveldimo nefrito genetinis pagrindas – IV tipo kolageno geno mutacija (Fiengold ir kt., 1985).
Genetinio ligos pobūdžio tyrimas leido mums daryti išvadą, kad paveldimo nefrito fenotipinių apraiškų skirtumai (su klausos praradimu ar be jo) atsiranda dėl mutantinio geno raiškos laipsnio. Taigi, šiuo metu visi klinikiniai variantai laikomi vienos ligos apraiškomis, o terminas „paveldimas nefritas“ yra sinonimas terminui „Alporto sindromas“.
Remiantis epidemiologiniais tyrimais, paveldimas nefritas pasireiškia 17 atvejų 100 000 vaikų.
 [
[ Alporto sindromo priežastys
Genetinis ligos pagrindas yra IV tipo kolageno α-5 grandinės geno mutacija. Šis tipas yra universalus inkstų, kochlearinio aparato, lęšiuko kapsulės, tinklainės ir ragenos pamatinėms membranoms, kas buvo įrodyta tyrimuose, kuriuose naudojami monokloniniai antikūnai prieš šią kolageno frakciją. Neseniai buvo nurodyta DNR zondų naudojimo prenatalinei paveldimo nefrito diagnostikai galimybė.
Pabrėžiama, kaip svarbu ištirti visus šeimos narius DNR zondais, siekiant nustatyti mutantinio geno nešiotojus, o tai labai svarbu atliekant medicininę ir genetinę konsultaciją šeimoms, sergančioms šia liga. Tačiau iki 20% šeimų neturi giminaičių, sergančių inkstų liga, o tai rodo didelį savaiminių nenormalaus geno mutacijų dažnį. Dauguma pacientų, sergančių paveldimu nefritu, savo šeimose turi asmenų, sergančių inkstų liga, klausos praradimu ir regėjimo patologija; svarbios yra kraujomaišos tarp žmonių, turinčių vieną ar daugiau protėvių, nes giminingų asmenų santuokose padidėja tikimybė gauti tuos pačius genus iš abiejų tėvų. Nustatyti autosominis dominantinis, autosominis recesyvinis ir dominantinis, su X chromosoma susiję perdavimo keliai.
Vaikams dažniausiai išskiriami trys paveldimo nefrito tipai: Alporto sindromas, paveldimas nefritas be klausos praradimo ir šeimyninė gerybinė hematurija.
Alporto sindromas yra paveldimas nefritas su klausos sutrikimu. Jis atsiranda dėl kombinuoto inkstų, ausies ir akių struktūrų glomerulų pamatinės membranos kolageno struktūros defekto. Klasikinio Alporto sindromo genas yra X chromosomos ilgosios rankos 21-22 q lokuse. Daugeliu atvejų jis paveldimas dominantiniu būdu, susijęs su X chromosoma. Šiuo atžvilgiu Alporto sindromas vyrams yra sunkesnis, nes moterims mutantinio geno funkciją kompensuoja sveikas antrosios, nepažeistos chromosomos alelis.
Paveldimojo nefrito išsivystymo genetinis pagrindas yra IV tipo kolageno alfa grandinių genų mutacijos. Yra žinomos šešios IV tipo kolageno G alfa grandinės: a5 ir a6 grandinių genai (Col4A5 ir Col4A5) yra X chromosomos ilgojoje rankoje 21-22q zonoje; a3 ir a4 grandinių genai (Col4A3 ir Col4A4) yra 2-oje chromosomoje; a1 ir a2 grandinių genai (Col4A1 ir Col4A2) yra 13-oje chromosomoje.
Daugeliu atvejų (80–85 %) nustatomas su X chromosoma susijęs ligos paveldėjimo modelis, susijęs su Col4A5 geno pažeidimu dėl ištrynimo, taškinių mutacijų arba splaisingo sutrikimų. Šiuo metu nustatyta daugiau nei 200 Col4A5 geno mutacijų, kurios sukelia IV tipo kolageno a5 grandinių sintezės sutrikimą. Esant tokiam paveldėjimo tipui, liga pasireiškia abiejų lyčių vaikams, tačiau berniukams ji yra sunkesnė.
Mutacijos Col4A3 ir Col4A4 genų, atsakingų už IV tipo kolageno a3 ir a4 grandinių sintezę, lokusuose paveldimos autosominiu būdu. Tyrimų duomenimis, autosominis dominantinis paveldėjimo tipas stebimas 16 % paveldimo nefrito atvejų, o autosominis recesyvinis – 6 % pacientų. Yra žinoma apie 10 Col4A3 ir Col4A4 genų mutacijų variantų.
Mutacijų rezultatas – IV tipo kolageno surinkimo procesų pažeidimas, dėl kurio pažeidžiama jo struktūra. IV tipo kolagenas yra vienas iš pagrindinių glomerulų pamatinės membranos, kochlearinio aparato ir akies lęšiuko komponentų, kurių patologija bus nustatyta paveldimo nefrito klinikoje.
IV tipo kolagenas, kuris yra glomerulų pamatinės membranos dalis, daugiausia susideda iš dviejų a1 grandinių (IV) ir vienos a2 grandinės (IV), taip pat turi a3, a4, a5 grandines. Dažniausiai, paveldint su X chromosoma, Col4A5 geno mutaciją lydi a3, a4, a5 ir a6 grandinių nebuvimas IV tipo kolageno struktūroje, o o1 ir a2 grandinių skaičius glomerulų pamatinėje membranoje padidėja. Šio reiškinio mechanizmas nėra aiškus, manoma, kad priežastis yra po transkripcijos vykstantys mRNR pokyčiai.
IV tipo kolageno, esančio glomerulų pamatinėse membranose, struktūroje esančių a3, a4 ir a5 grandinių nebuvimas lemia jų plonėjimą ir trapumą ankstyvosiose Alporto sindromo stadijose, kuris kliniškai dažniau pasireiškia hematurija (rečiau hematurija su proteinurija arba tik proteinurija), klausos praradimu ir lentikonusu. Tolesnė ligos progresavimas vėlyvosiose stadijose lemia pamatinių membranų sustorėjimą ir pralaidumo sutrikimą, jose dauginasi V ir VI tipo kolagenas, pasireiškiantis proteinurijos padidėjimu ir inkstų funkcijos sumažėjimu.
Paveldimo nefrito fone esančios mutacijos pobūdis daugiausia lemia jo fenotipinę pasireiškimą. X chromosomos ištrynimo atveju, kai kartu mutuoja ir Col4A5 bei Col4A6 genai, atsakingi už IV tipo kolageno a5 ir a6 grandinių sintezę, Alporto sindromas derinamas su stemplės ir lytinių organų lejomiomatoze. Tyrimų duomenimis, esant su ištrynimu susijusiai Col4A5 geno mutacijai, pastebimas didesnis patologinio proceso sunkumas, inkstų pažeidimo derinys su ekstrarenalinėmis apraiškomis ir ankstyva lėtinio inkstų nepakankamumo išsivystymo stadija, palyginti su šio geno taškine mutacija.
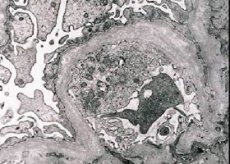
Morfologiškai elektroninė mikroskopija atskleidžia glomerulų pamatinių membranų (ypač lamina densa) suplonėjimą ir stratifikaciją bei elektronų tankių granulių buvimą. Glomerulų pažeidimai tame pačiame paciente gali būti nevienodi – nuo minimalių židininių mezangialinių pažeidimų iki glomerulosklerozės. Alporto sindromo glomerulitas visada yra imunoneigiamas, tuo jis skiriasi nuo glomerulonefrito. Būdingi požymiai yra kanalėlių atrofijos išsivystymas, limfohistiocitinė infiltracija ir „putų ląstelių“ su lipidų intarpais – lipofagų – buvimas. Ligai progresuojant, atskleidžiamas glomerulų pamatinių membranų sustorėjimas ir ryškus jų irimas.
Pastebimi tam tikri imuninės sistemos pokyčiai. Sergant paveldimu nefritu, sumažėja Ig A kiekis ir padidėja IgM koncentracija kraujyje, ankstyvosiose ligos stadijose IgG kiekis gali būti padidėjęs, o vėlyvosiose – sumažėti. Galbūt IgM ir G koncentracijos padidėjimas yra savotiška kompensacinė reakcija į IgA trūkumą.
Sumažėja T limfocitų sistemos funkcinis aktyvumas; pastebimas selektyvus B limfocitų, atsakingų už Ig A sintezę, sumažėjimas, sutrinka imuniteto fagocitinis ryšys, daugiausia dėl chemotaksės ir tarpląstelinių virškinimo procesų sutrikimo neutrofiluose.
Tiriant inkstų biopsiją pacientams, sergantiems Alporto sindromu, elektroninės mikroskopijos duomenys atskleidžia ultrastruktūrinius glomerulų pamatinės membranos pokyčius: glomerulų pamatinės membranos retėjimą, struktūros sutrikimą ir skilimą, pasikeitus jos storiui ir nelygiems kontūrams. Ankstyvosiose paveldimo nefrito stadijose defektas lemia glomerulų pamatinių membranų retėjimą ir trapumą.
Glomerulų membranų suplonėjimas yra palankesnis požymis ir dažniau pasireiškia mergaitėms. Pastovesnis elektronmikroskopinis paveldimo nefrito požymis yra pamatinės membranos įskilimas, o jos pažeidimo sunkumas koreliuoja su proceso sunkumu.
Alporto sindromo simptomai vaikams
Pirmieji Alporto sindromo simptomai, pasireiškiantys izoliuotu šlapimo sindromu, dažniausiai nustatomi pirmųjų trejų gyvenimo metų vaikams. Daugeliu atvejų liga nustatoma atsitiktinai. Šlapimo sindromas nustatomas profilaktinio vaiko tyrimo metu, prieš priimant į vaikų priežiūros įstaigą arba ŪRI metu. Jei ŪRI metu šlapime aptinkama patologija. Sergant paveldimu nefritu, skirtingai nei įgytu glomerulonefritu, latentinio periodo nėra.
Pradinėje ligos stadijoje vaiko sveikata nukenčia nedaug, būdingas bruožas yra šlapimo sindromo užsitęsimas ir atsparumas. Vienas iš pagrindinių požymių yra įvairaus sunkumo hematurija, stebima 100% atvejų. Hematurijos laipsnio padidėjimas pastebimas kvėpavimo takų infekcijų, fizinio krūvio metu arba po jų, po profilaktinių skiepų. Proteinurija daugeliu atvejų neviršija 1 g per parą, ligos pradžioje gali būti nepastovi, procesui progresuojant, proteinurija didėja. Periodiškai šlapimo nuosėdose gali būti leukociturija, kurioje vyrauja limfocitai, o tai susiję su intersticinių pokyčių atsiradimu.
Vėliau sutrinka dalinė inkstų funkcija, pablogėja bendra paciento būklė: atsiranda intoksikacija, raumenų silpnumas, arterinė hipotenzija, dažnai klausos sutrikimas (ypač berniukams), o kartais ir regos sutrikimai. Intoksikacija pasireiškia blyškumu, nuovargiu ir galvos skausmais. Pradinėje ligos stadijoje klausos praradimas daugeliu atvejų nustatomas tik atlikus audiografiją. Klausos praradimas sergant Alporto sindromu gali pasireikšti įvairiais vaikystės laikotarpiais, tačiau dažniausiai klausos praradimas diagnozuojamas 6–10 metų amžiuje. Vaikų klausos praradimas prasideda nuo aukštų dažnių, pasiekiant reikšmingą oro ir kaulų laidumo laipsnį, pereinant nuo garsą laidžiančio iki garsą suvokiančio klausos praradimo. Klausos praradimas gali būti vienas pirmųjų ligos simptomų ir gali pasireikšti prieš šlapimo sindromą.
20 % atvejų pacientams, sergantiems Alporto sindromu, pasireiškia regos organų pokyčiai. Dažniausiai aptinkamos lęšiuko anomalijos: sferofokija, priekinis, užpakalinis arba mišrus lęšiukonusas ir įvairios kataraktos. Šeimose, sergančiose Alporto sindromu, pastebimas didelis trumparegystės dažnis. Nemažai tyrėjų šiose šeimose nuolat pastebi dvišalius perimakulinius pokyčius ryškiai balkšvų arba gelsvų granuliacijų geltonkūnyje pavidalu. Jie šį požymį laiko nuolatiniu simptomu, turinčiu didelę diagnostinę vertę sergant Alporto sindromu. K. S. Chugh ir kt. (1993) oftalmologiniame tyrime pacientams, sergantiems Alporto sindromu, 66,7 % atvejų sumažėjo regėjimo aštrumas, 37,8 % – priekinis lęšiukonusas, 22,2 % – tinklainės dėmės, 20 % – katarakta ir 6,7 % – keratokonusas.
Kai kuriems vaikams, sergantiems paveldimu nefritu, ypač išsivystius inkstų nepakankamumui, pastebimas reikšmingas fizinio vystymosi atsilikimas. Inkstų nepakankamumui progresuojant, išsivysto arterinė hipertenzija. Vaikams ji dažniau nustatoma paauglystėje ir vyresnio amžiaus grupėse.
Paveldimu nefritu sergantiems pacientams būdingos įvairios (daugiau nei 5–7) jungiamojo audinio dismorfogenezės stigmos. Tarp pacientų jungiamojo audinio stigmų dažniausiai pasitaiko akių hipertelorizmas, aukštas gomurys, sąkandžio anomalijos, nenormali ausų forma, mažojo piršto išlinkimas ant rankų ir „sandalo tarpas“ ant kojų. Paveldimam nefritui būdingas dismorfogenezės stigmų vienodumas šeimoje, taip pat didelis jų paplitimo dažnis tarp probandų, kurių linija perduodama liga, giminaičių.
Ankstyvosiose ligos stadijose nustatomas izoliuotas dalinių inkstų funkcijų sumažėjimas: aminorūgščių, elektrolitų pernašos, koncentracijos funkcijos, acidogenezės, vėlesni pokyčiai veikia tiek proksimalinės, tiek distalinės nefrono dalių funkcinę būklę ir pasižymi kombinuotais daliniais sutrikimais. Glomerulų filtracijos sumažėjimas pasireiškia vėliau, dažniau paauglystėje. Paveldimam nefritui progresuojant, vystosi anemija.
Taigi, paveldimas nefritas pasižymi laipsniška ligos eiga: pirmiausia latentinė stadija arba paslėpti klinikiniai simptomai, pasireiškiantys minimaliais šlapimo sindromo pokyčiais, vėliau vyksta laipsniška proceso dekompensacija, sumažėja inkstų funkcija su akivaizdžiais klinikiniais simptomais (intoksikacija, astenija, vystymosi sulėtėjimas, anemija). Klinikiniai simptomai paprastai pasireiškia nepriklausomai nuo uždegiminės reakcijos sluoksniavimo.
Paveldimas nefritas gali pasireikšti skirtingu amžiumi, priklausomai nuo geno, kuris iki tam tikro laiko yra represuotoje būsenoje, veikimo.
Klasifikacija
Yra trys paveldimo nefrito tipai
- I variantas – kliniškai pasireiškia kaip nefritas su hematurija, klausos praradimu ir akių pažeidimu. Nefrito eiga progresuoja, vystosi lėtinis inkstų nepakankamumas. Paveldėjimo tipas yra dominuojantis, susijęs su X chromosoma. Morfologiškai išryškėja pamatinės membranos struktūros pažeidimas, jos plonėjimas ir skilimas.
- II variantas – kliniškai pasireiškia kaip nefritas su hematurija be klausos praradimo. Nefrito eiga progresuoja, vystosi lėtinis inkstų nepakankamumas. Paveldėjimo tipas dominuojantis, susijęs su X chromosoma. Morfologiškai nustatomas glomerulų kapiliarų pamatinės membranos (ypač laminadensos) suplonėjimas.
- III variantas – gerybinė šeiminė hematurija. Eiga palanki, lėtinis inkstų nepakankamumas neišsivysto. Paveldėjimo tipas – autosominis dominantinis arba autosominis recesyvinis. Esant autosominiam recesyviniam paveldėjimo tipui, moterims pastebima sunkesnė ligos eiga.
Alporto sindromo diagnozė
Siūlomi šie kriterijai:
- bent dviejų pacientų, sergančių nefropatija, buvimas kiekvienoje šeimoje;
- hematurija kaip pagrindinis nefropatijos simptomas probande;
- klausos praradimo buvimas bent vienam šeimos nariui;
- lėtinio inkstų nepakankamumo išsivystymas vienam ar keliems giminaičiams.
Diagnozuojant įvairias paveldimas ir įgimtas ligas, didelis dėmesys skiriamas visapusiškam požiūriui į tyrimą ir, svarbiausia, dėmesiu duomenims, gautiems sudarant vaiko kilmės knygą. Alporto sindromo diagnozė laikoma pagrįsta tais atvejais, kai pacientui nustatomi 3 iš 4 tipinių požymių: hematurija ir lėtinis inkstų nepakankamumas šeimoje, neurosensorinis klausos praradimas, regėjimo patologija pacientui, glomerulų pamatinės membranos skilimo požymių nustatymas su jos storio pokyčiais ir nelygiais kontūrais atliekant elektroninės mikroskopijos biopsijos charakteristikas.
Paciento tyrimas turėtų apimti klinikinius ir genetinius tyrimo metodus; tikslinį ligos istorijos tyrimą; bendrą paciento tyrimą, atsižvelgiant į diagnostiniu požiūriu reikšmingus kriterijus. Kompensacijos stadijoje patologiją galima nustatyti tik sutelkiant dėmesį į tokius sindromus kaip paveldimas sutrikimas, hipotenzija, daugybinės disembriogenezės stigmos, šlapimo sindromo pokyčiai. Dekompensacijos stadijoje gali pasireikšti ekstrarenaliniai simptomai, tokie kaip sunkus apsinuodijimas, astenija, uždelstas fizinis vystymasis, anemija, pasireiškiantys ir sustiprėjantys palaipsniui mažėjant inkstų funkcijai. Daugumai pacientų, mažėjant inkstų funkcijai, stebima: sumažėjusi acidogenezė ir aminogenezė; 50% pacientų pastebi reikšmingą inkstų sekrecinės funkcijos sumažėjimą; ribotą šlapimo optinio tankio svyravimų diapazoną; filtracijos ritmo sutrikimą, o vėliau ir glomerulų filtracijos sumažėjimą. Lėtinio inkstų nepakankamumo stadija diagnozuojama, kai pacientų kraujo serume 3–6 mėnesius ar ilgiau yra padidėjęs šlapalo kiekis (daugiau nei 0,35 g/l), o glomerulų filtracija sumažėja iki 25 % normos.
Paveldimojo nefrito diferencinė diagnostika pirmiausia turėtų būti atliekama esant hematurinei įgyto glomerulonefrito formai. Įgytas glomerulonefritas dažniausiai prasideda ūmiai, praėjus 2–3 savaitėms po infekcijos, pasireiškia ekstrarenaliniais požymiais, įskaitant hipertenziją nuo pirmųjų dienų (paveldimojo nefrito atveju, priešingai – hipotenzija), sumažėjusia glomerulų filtracija ligos pradžioje, dalinių kanalėlių funkcijų sutrikimų nėra, o paveldimo nefrito atveju jie yra. Įgytas glomerulonefritas pasireiškia ryškesne hematurija ir proteinurija, padidėjusiu ESR. Diagnostinę vertę turi tipiški glomerulų pamatinės membranos pokyčiai, būdingi paveldimam nefritui.
Diferencinė dismetabolinės nefropatijos diagnostika atliekama esant lėtiniam inkstų nepakankamumui, šeimoje kliniškai nustatytoms heterogeninėms inkstų ligoms, ir gali būti nefropatijos spektras nuo pielonefrito iki urolitiazės. Vaikai dažnai skundžiasi pilvo skausmu ir periodiškai šlapinantis, šlapimo nuosėdose – oksalatais.
Įtarus paveldimą nefritą, pacientas turėtų būti nukreiptas į specializuotą nefrologijos skyrių diagnozei patikslinti.
Ką reikia išnagrinėti?
Kokie testai reikalingi?
Su kuo susisiekti?
Alporto sindromo gydymas
Režimas apima didelio fizinio krūvio ir gryno oro buvimo apribojimus. Mityba yra pilnavertė, pakankamas pilnaverčių baltymų, riebalų ir angliavandenių kiekis, atsižvelgiant į inkstų funkciją. Labai svarbu nustatyti ir gydyti lėtinius infekcijos židinius. Vartojami šie vaistai: ATP, kokarboksilazė, piridoksinas (iki 50 mg/d.), karnitino chloridas. Kursai skiriami 2–3 kartus per metus. Hematurijai gydyti skiriami vaistažolių preparatai – dilgėlė, aronijų sultys, kraujažolė.
Užsienio ir šalies literatūroje yra pranešimų apie gydymą prednizolonu ir citostatikų vartojimą. Tačiau sunku įvertinti jų poveikį.
Lėtinio inkstų nepakankamumo atveju naudojama hemodializė ir inkstų transplantacija.
Specifinių (veiksmingų patogenetinių) paveldimo nefrito gydymo metodų nėra. Visos gydymo priemonės skirtos inkstų funkcijos silpnėjimo prevencijai ir sulėtinimui.
Mityba turėtų būti subalansuota ir kaloringa, atsižvelgiant į inkstų funkcinę būklę. Nesant funkcinių sutrikimų, vaiko mityboje turėtų būti pakankamai baltymų, riebalų ir angliavandenių. Esant inkstų funkcijos sutrikimo požymiams, reikia riboti baltymų, angliavandenių, kalcio ir fosforo kiekį, o tai atitolina lėtinio inkstų nepakankamumo vystymąsi.
Fizinis aktyvumas turėtų būti ribotas; vaikams patariama vengti sporto.
Reikėtų vengti kontakto su infekciniais pacientais, sumažinti ūminių kvėpavimo takų ligų išsivystymo riziką. Būtina sanitarija lėtinės infekcijos židiniuose. Profilaktinės vakcinacijos vaikams, sergantiems paveldimu nefritu, neatliekamos, vakcinacija galima tik esant epidemiologinėms indikacijoms.
Hormoninis ir imunosupresinis gydymas paveldimo nefrito atveju yra neefektyvus. Yra požymių, kad ilgalaikis, daugelį metų vartojant ciklosporiną A ir AKF inhibitorius, pastebimas teigiamas poveikis (proteinurijos sumažėjimas ir ligos progresavimo sulėtėjimas).
Pacientų gydymui naudojami vaistai, kurie gerina medžiagų apykaitą:
- piridoksinas – 2–3 mg/kg/d. 3 dozėmis 4 savaites;
- kokarboksilazė - 50 mg į raumenis kas antrą dieną, iš viso 10-15 injekcijų;
- ATP - 1 ml į raumenis kas antrą dieną, 10-15 injekcijų;
- vitaminas A – 1000 TV/metus/dieną 1 doze 2 savaites;
- Vitaminas E – 1 mg/kg/d., 1 doze, 2 savaites.
Šis gydymo būdas padeda pagerinti bendrą pacientų būklę, sumažinti kanalėlių disfunkciją ir atliekamas kursais 3 kartus per metus.
Levamizolis gali būti vartojamas kaip imunomoduliatorius – 2 mg/kg/dieną 2–3 kartus per savaitę, tarp dozių darant 3–4 dienų pertraukas.
Remiantis tyrimų duomenimis, hiperbarinė oksigenacija teigiamai veikia hematurijos ir inkstų funkcijos sutrikimo sunkumą.
Veiksmingiausias paveldimo nefrito gydymo metodas yra savalaikė inkstų transplantacija. Šiuo atveju transplantacijos metu liga neatsinaujina; nedideliu procentu atvejų (apie 5 %) persodintame inkste gali išsivystyti nefritas, susijęs su glomerulų pamatinės membranos antigenais.
Perspektyvi kryptis yra prenatalinė diagnostika ir genų inžinerijos terapija. Eksperimentai su gyvūnais rodo didelį normalių genų, atsakingų už IV tipo kolageno alfa grandinių sintezę, perkėlimo į inkstų audinį efektyvumą, po kurio stebima normalių kolageno struktūrų sintezė.
Prognozė
Paveldimojo nefrito prognozė visada yra rimta.
Prognoziškai nepalankūs paveldimo nefrito eigos kriterijai yra šie:
- vyriška lytis;
- ankstyvas lėtinio inkstų nepakankamumo vystymasis šeimos nariams;
- proteinurija (daugiau nei 1 g per dieną);
- glomerulų bazinių membranų sustorėjimas mikroskopu;
- akustinis neuritas;
- ištrynimas Col4A5 gene.
Gerybinės šeiminės hematurijos prognozė yra palankesnė.
Использованная литература