Medicinos ekspertas

Naujos publikacijos
Paveldimas nefritas (Alporto sindromas) vaikams
Paskutinį kartą peržiūrėta: 23.04.2024

Visas „iLive“ turinys yra peržiūrėtas medicinoje arba tikrinamas, kad būtų užtikrintas kuo didesnis faktinis tikslumas.
Mes turime griežtas įsigijimo gaires ir susiejamos tik su geros reputacijos žiniasklaidos svetainėmis, akademinių tyrimų institucijomis ir, jei įmanoma, medicininiu požiūriu peržiūrimais tyrimais. Atkreipkite dėmesį, kad skliausteliuose ([1], [2] ir tt) esantys numeriai yra paspaudžiami nuorodos į šias studijas.
Jei manote, kad bet koks mūsų turinys yra netikslus, pasenęs arba kitaip abejotinas, pasirinkite jį ir paspauskite Ctrl + Enter.
Paveldima nefritas (alport sindromas) - genetiškai lemia ne imuninė paveldėjo glomerulopatiją eksponuoti hematurija (kartais proteinurija), progresuojantis inkstų funkcijos lėtiniu inkstų nepakankamumu plėtros yra dažnai susijęs su neurosensorinis kurtumas ir silpnaregiams.
Pirmą kartą šią ligą 1902 m. Apibūdino LGGuthrie, kuris pastebėjo kelių kartų šeimą, kuriam stebėta hematurija. 1915 m. Tos pačios AFHurst šeimos nariai apibūdino uremos vystymąsi. 1927 m. "Alport" pirmą kartą nustatė kurtumą kai kuriuose giminaičiuose su hematurija. Praėjusio amžiaus penkiasdešimtmečio metu tokia liga buvo aptikta akių sužalojimų. 1972 m. Pacientams, kuriems buvo paveldima hematurija, morfologiškai tiriant inkstų audinius, Hinglais ir kt. Atskleidė netolygus glomerulinių bazalinių membranų išsiplėtimas ir dilatinimas. 1985 m. Nustatytas paveldimo nefrito genetinis pagrindas - IV tipo kolageno geno mutacija (Fiengold et al., 1985).
Ligos genetinio pobūdžio tyrimas leido daryti išvadą, kad paveldimojo nefrito fenotipinių pasireiškimų skirtumai (su klausos praradimu ar be jo) priklauso nuo mutantinio geno ekspresijos laipsnio. Taigi, šiuo metu visi klinikiniai variantai yra laikomi vienos ligos požymiais, o terminas "paveldimasis nefritas" yra sinonimas su terminu "Alporto sindromas".
Remiantis epidemiologiniais tyrimais, paveldimasis nefritas atsiranda dažniau kaip 17 iš 100 000 vaikų.
Alporto sindromo priežastys
Genetinis ligos pagrindas yra IV tipo kolageno grandinės geno a-5 mutacija. Šis universalus bazinių inkstų membranų, kochleariniam prietaiso, lęšių kapsulių, tinklainės ir ragenos tipo, kad parodyta tyrimai, naudojant monokloninius antikūnus prieš kolageno frakcija. Neseniai jie nurodo galimybę naudoti DNR zondus, skirtus paveldimojo nefrito prenataliniam diagnozavimui.
Pabrėžiama, kaip svarbu išbandyti visus šeimos narius, naudojant DNR zondus, norint nustatyti mutantinio geno nešėjus, o tai labai svarbu atliekant medicininę genetinę konsultaciją šeimoms, turinčioms šią ligą. Tačiau iki 20% šeimų neturi giminaičių su inkstų liga, o tai rodo didelį spontaninių mutacijų paplitimą nenormalų genų. Dauguma pacientų, kurių šeimoje yra paveldimas nefritas, yra asmenys, serganti inkstų liga, klausos praradimu ir regos patologija; susijusių su viena ar daugiau protėvių turinčių žmonių santuokų, nes susijusių asmenų santuoka padidina tikimybę gauti tuos pačius genus iš abiejų tėvų. Sukurta autosominė dominuojanti ir autosominė recesinė ir dominuojanti, susieta su perdavimo tako X chromosoma.
Vaikai labiau linkę atskirti tris paveldimo nefrito variantus: Alporto sindromą, paveldimą nefritą be klausos praradimo ir šeimos nepalankiąją hematuriją.
Alporto sindromas - paveldimas nefritas su klausos pažeidimu. Pagrindas yra sujungtų inkstų glomerulų bazinės membranos kolageno struktūros defektas, ausų ir akių struktūros. Klasikinio Alporto sindromo genas yra ties X-chromosomos ilgosios rankos lokusu 21-22 q. Daugeliu atvejų ji yra paveldima dominuojančio tipo, susieto su X chromosoma. Šiuo atžvilgiu vyrams Alporto sindromas yra sunkesnis, nes moterims mutantinė geno funkcija kompensuojama sveika alelio antroji, nepažeista chromosoma.
Paveldimo nefrito vystymosi genetinis pagrindas yra IV tipo kolageno alfa grandžių genų mutacijos. Jis yra žinomas kaip šešių-grandines IV tipo kolageno G: A5 ir A6 genų tinklai (Sol4A5 ir Sol4A5) yra ant ilgojo peties X chromosomos į 21-22q zonoje; a3- ir a4-grandinių genai (Co4A3 ir Co4A4) - ant 2-osios chromosomos; a1- ir a2-grandinių genai (Co4A1 ir Co4A2) - 13 chromosomos.
Daugeliu atvejų (80-85%) su X susijusi ligos paveldėjimo rūšis yra susijusi su Co4A5 geno pažeidimu dėl ištrynimo, taškų mutacijų ar susiliejimo sutrikimų. Šiuo metu randama daugiau kaip 200 geno Kol4A5 mutacijų, atsakingų už IV tipo kolageno a5 grandinių sintezės pažeidimą. Tokio paveldėjimo atveju liga pasireiškia abiejų lyčių vaikais, bet berniukuose tai sunkiau.
Gautų Co4A3 ir Co4A4 genų mutacijos, atsakingos už IV tipo kolageno a3 ir a4 grandinių sintezę, yra paveldimos autosomely. Remiantis tyrimais, 16 proc. Atvejų paveldimų nefritų, autosominių recesyvinių - 6 proc. Pacientų, stebimas autosominis dominuojantis paveldėjimo tipas. Yra apie 10 Co4A3 ir Co4A4 genų mutacijų.
Dėl mutacijų rezultatas yra IV tipo kolageno surinkimo procesų pažeidimas, dėl kurio sutrinka jo struktūra. IV tipo kolagenas yra viena iš pagrindinių glomerulų bazės membranos, kochlerio aparato ir akies lęšio, kurios patologija atskleidžiama paveldimo nefrito klinikoje, komponentai.
Kolageno IV tipo, dalis glomerulų pamatinės membranos, iš esmės susideda iš dviejų grandinės a1 (IV) ir vienas A2 grandinės (IV), ir taip pat yra A3, A4, A5 grandinę. Dažniausiai, kai X chromosoma susijusi paveldėjimo Sol4A5 mutacija lydi trūksta A3, A4 ir A6 A5 grandinės kolageno IV tipo struktūros ir O1 ir A2 grandinių skaičius į glomerulų pamatinės membranos didėja. Šio reiškinio mechanizmas yra neaiškus, daroma prielaida, kad priežastis yra postrekūniniai pokyčiai mRNR.
Trūksta A3, A4 ir A5 grandinės struktūros IV tipo kolageno pamatinės membranos iš glomerulų rezultatų ugdymo ir silpnumo ankstyvosiose stadijose Alport sindromu, kuris pasireiškia kliniškai dauguma hematurija (kartais hematurija ar proteinurija tik proteinurija), klausos ir lenticonus. Toliau ligos progresavimą veda prie sustorėjimo, gausumas ir pamatinės membranos pralaidumas pabaigoje ligos stadijose, su šių rūšių kolageną V ir VI, pasireikštų proteinurijos ir sumažintų inkstų funkciją augimo.
Paveldimo nefrito pagrindu vykstančios mutacijos pobūdis labiausiai lemia jo fenotipinį pasireiškimą. Kai X chromosomų Išbrauktos su tuo pat metu mutacijų ir Sol4A6 Sol4A5 genų, atsakingų už A5 ir A6 grandines IV tipo kolageno sintezės, kartu su alport sindromui leiomyomatosis stemplės ir lytinių organų. Remiantis tyrimais su Sol4A5 genų mutacijų, susijusių su ištrynimo yra pažymėtos didelis sunkumas patologinio proceso, derinys su inkstų pažeidimo extrarenal apraiškų ankstyvoje vystymosi lėtinio inkstų funkcijos nepakankamumo, palyginti stochechnoy mutacija šio geno.
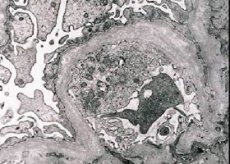
Morfologiškai elektronų mikroskopija atskleidžia glomerulų bazalinių membranų (ypač lamina densa) sulėtėjimą ir atskyrimą ir elektroniškai tankių granulių buvimą. Glomerulų pažeidimas tame pačiame paciente gali būti nevienodas, nuo mažiausio židininio mezangio pažeidimo iki glomerulosklerozės. Alporto sindromas glomerulitas visada yra imuninės neigiamas, kuris išskiria jį nuo glomerulonefrito. Būdinga tai kanalo atrofijos, limfichistocitų infiltracijos, "putų ląstelių" buvimas su lipidų - lipofagų įtraukimu. Su ligos progresavimu atskleidžiamas bazinių glomerulų membranų storėjimas ir ryškus sunaikinimas.
Atskleidžiami tam tikri imuninės sistemos būklės pokyčiai. Pacientams, kuriems yra paveldimas nefritas, pastebimas IgA lygio sumažėjimas ir tendencija didinti IgM koncentraciją kraujyje, IgG lygis gali būti padidintas ankstyvose ligos vystymosi stadijose ir vėlyvuose laikotarpiuose. Galbūt padidėjusi IgM ir G koncentracija yra kompensacinis atsakas į IgA trūkumą.
T-limfocitų sistemos funkcinis aktyvumas mažėja; Ji pažymėta selektyviai sumažinant B-limfocitų, atsakingas už Ig A sintezės, suskirstytas, fagocitozės imunitetą nuorodą, daugiausia dėl to, suskirstymą procesų chemotaksio ir viduląstelinio virškinimo neutrofilų
Dėl inkstų biopsijos tyrimo pacientams, sergantiems Alport sindromo elektroniniu mikroskopu, ULTRASTRUKTŪRINIAI pakitimai glomerulų prieš bazinę membraną: retinimo, ir išskirstomi modelius apie pažeidimus glomerulų pamatinės membranas su jos storio ir nelygių kontūrų kaita. Ankstyvosiose paveldimiausio nefrito stadijose defektas nustato glomerulinių bazinių membranų retinimą ir trapumą.
Glomerulinių membranų išsilyginimas yra palankesnis ženklas ir dažniau pasireiškia merginoms. Paveldėtas nefritas yra labiau pastovus elektronų mikroskopinis požymis - bazinės membranos skilimas, o jo sunaikinimo sunkumas koreliuoja su proceso sunkumu.

Alporto sindromo simptomai vaikams
Pirmieji Alporto sindromo simptomai izoliuoto šlapimo sindromo forma dažniau nustatomi pirmųjų trejų metų gyvenimo vaikams. Daugeliu atvejų liga nustatoma atsitiktinai. Šlapimo sindromas atsiskleidžia atliekant prevencinį vaiko tyrimą prieš įvažiuojant į vaikų įstaigą ar ARVI. Jei pasireiškia patologija šlapime per ARVI. Paveldintam nefritui, kitaip nei įgytas glomerulonefritas, nėra latentinio laikotarpio.
Pradinėje ligos stadijoje vaiko gerovė kenčia mažai, būdinga šlapimo sindromo patvarumai ir ištvermė. Vienas iš pagrindinių požymių yra įvairios laipsnio hematurija, pastebėta 100% atvejų. Hematurijos laipsnio padidėjimas pastebimas kvėpavimo takų infekcijų metu, po fizinio krūvio ar po profilaktinio skiepijimo. Dauguma atvejų proteinuuras neviršija 1 g per parą, ligos pradžioje gali būti nestabilios, nes progresuojant progresuoja proteinurija. Periodiškai šlapimo nuosėdose gali būti leukociturija su limfocitų dominavimu, kuri yra susijusi su intersticinių pokyčių atsiradimu.
Daugiau yra dalinio Pablogėjus inkstų funkcijai bendrosios ligonio būklę pažeidimas: ten intoksikacijos, raumenų silpnumas, hipotenzija, dažnai klausos praradimas (ypač berniukams) kartais neryškus matymas. Toksikacija pasireiškia blyškiu, nuovargiu, galvos skausmu. Pradiniame ligos stadijoje klausos praradimas dažniausiai nustatomas tik audiogrāfija. Alporto sindromo klausos praradimas gali pasireikšti įvairiais vaikystės laikais, tačiau dažniausiai klausos netekimas diagnozuojamas 6-10 metų amžiaus. Klausos praradimas vaikams prasideda dideliais dažniais, daugiausia pasiekus ore ir kaulų laidumą, pereinant nuo garso laidumo iki garso perduodančio klausos praradimo. Klausos praradimas gali būti vienas iš pirmųjų ligos simptomų ir gali būti anksčiau už šlapimo sindromą.
20% atvejų pacientams, sergantiems Alporto sindromu, pasikeičia akys. Dažniausiai aptikta anomalija nuo lęšio: sferofokiya, lenticonus priekiniai, galiniai arba mišri, iš katarakta įvairovė. Šeimose, turinčiose Alporto sindromą, pastebimas trumparegystė. Tyrėjų skaičius yra nuolat Šios šeimos švęsti dvišalius perimakulyarnye pakeitimus kaip ryškių balkšvų arba gelsvų granuliacijos į geltonkūnio. Jie laiko šį simptomą nuolatiniu simptomu, kuris turi didelę diagnostiką Alporto sindromu. C. S. Chugh ir kt. (1993) ir oftalmologiškai tyrimo metu Alport sindromas pacientams sumažėjo regos aštrumas yra 66,7% atvejų, pirmyn lenticonus - 37,8%, dėmės ant tinklainės - in 22,2%, katarakta - 20%, keratokonuso - 6 , 7%.
Kai kuriems paveldimiems nefritams vaikams, ypač inkstų nepakankamumo formavimui, pastebimas didelis fizinio vystymosi atsilikimas. Kadangi inkstų nepakankamumo progresavimo procese išsivysto hipertenzija. Vaikams tai dažniau nustatoma paauglystėje ir vyresnio amžiaus grupėse.
Būdinga tai, kad pacientams, turintiems įvairios (daugiau kaip 5-7) jungiamojo audinio stiebų dismembrogeno stigmos, yra paveldimas nefritas. Tarp jungiamojo audinio stigma pacientams su dažniausią akių hypertelorism, aukštos gomurio, netaisyklingas sąkandis, nenormalus formos ausis, kad pirštas ant rankų ", sandalevidnaya spragą" ant kojų kreivumas. Dėl paveldimos nefritas pasižymi vienodumas dizembriogeneza gėdos šeimoje, taip pat aukšto dažnio jų pasiskirstymą probands giminių, per kurį liga perduodama.
Pradžioje ligos stadijose atskleidė izoliuotą sumažinti dalinio inkstų funkcijos: transporto amino rūgščių, elektrolitų, koncentracijos funkcijų Acidogenesis, tolesni pakeitimai yra funkcinė būklė tiek proksimalinių ir distalinių nephron ir turi kombinuoto dalinių sutrikimų charakterį. Glomerulų filtracijos mažinimas atsiranda vėliau, dažniau paauglių laikotarpiu. Kai pasireiškia paveldimas nefritas, išsivysto anemija.
Tokiu būdu, paveldimas nefritas būdingas sustojimo ligos: pirmą latentinės etape arba paslėptų klinikinių simptomų pasireiškia nedidelius pokyčius šlapimo pūslės sindromo tada įvyksta laipsnišką dekompensacijos procesą su inkstų funkcijos sumažinimo su akivaizdžių klinikinių simptomų (intoksikacijos, astenija, vystymosi sutrikimų, anemizatsiya). Klinikiniai simptomai paprastai atsiranda nepriklausomai nuo uždegiminio atsako sluoksnių.
Paveldimasis nefritas gali pasireikšti skirtingais amžių laikotarpiais, kuris priklauso nuo geno veikimo, kuris iki tam tikro laiko yra represuotoje valstybėje.
Klasifikacija
Yra trys paveldimo nefrito variantai
- I variantas - kliniškai pasireiškia nefritu su hematurija, klausos praradimu ir akių pažeidimu. Nefrito eiga yra progresyvi, kai vystosi kreatinino klirensas. Paveldėjimo tipas yra dominuojantis, susietas su X chromosoma. Morfologiškai yra bazinės membranos struktūros trikdymas, jo retinimas ir skilimas.
- II variantas - kliniškai pasireiškia nefritu su hematurija be klausos praradimo. Nefrito eiga yra progresuojanti, kai pasireiškia lėtinis inkstų nepakankamumas. Paveldėjimo tipas yra dominuojantis, susietas su X chromosoma. Morfologiškai atskleidžiamas glomerulų kapiliarų bazinės membranos (ypač laminadensos) nykimas.
- III variantas - gerybinė šeimos hematurija. Kursas yra palankus, lėtinis inkstų nepakankamumas nėra išsivystęs. Paveldėjimo tipas yra autosominis dominuojantis arba autosominis recesyvinis. Autosominės recesyvinės paveldėjimo rūšys moterys turi sunkesnes ligos eigą.
Alporto sindromo diagnozė
Siūlomi šie kriterijai:
- kiekvienos šeimos buvimas bent dviem pacientams, sergantiems nefropatija;
- hematurija kaip pagrindinis nefropatijos simptomas probando;
- bent vienas šeimos narys turi klausos sutrikimą;
- lėtinio inkstų nepakankamumo vystymasis viename giminaite ir dar daugiau.
Jeigu paveldimų ir įgimtų ligų įvairovė diagnostikos svarbi vieta priklauso integruoto požiūrio į patikrinimo ir visų pirma atkreipti dėmesį į gautus duomenis į vaiko kilmės preparato. Diagnozė sindromas alport laikoma galioja tais atvejais, kai pacientas 3 iš 4 tipiškų bruožų: šeimoje hematurija ir lėtiniu inkstų nepakankamumu buvimas, iš paciento neurosensorinis klausos praradimą buvimas, patologijos radimo elektronų mikroskopiniai apibūdinimą biopsijos ženklų skilimo glomerulų bazinę membraną jos storio pasikeitimo ir netolygus kontūras.
Paciento tyrimas turėtų apimti klinikinius genetinius tyrimo metodus; nukreiptas ligos anamnezės tyrimas; Bendras paciento tyrimas, atsižvelgiant į diagnostinius kriterijus. Kompensacija etapas patologija gali sugauti tik sutelkiant dėmesį į tokius sindromus kaip turintys šeimos istoriją, hipotenzija, kelis purkas dizembriogeneza keičia šlapimo pūslės sindromas. Be dekompensuota estrarenalnyh gali sukelti tokius simptomus kaip stiprus apsvaigimas, astenija, atsilikusi fizinį vystymąsi anemizatsiya pasireiškia ir išplečianti su laipsniško mažėjimo inkstų funkciją. Daugumai pacientų, kuriems yra inkstų funkcijos sutrikimas, pastebimas rūgščių ir aminogenezės sumažėjimas; 50% pacientų pastebi žymį inkstų sekrecijos funkcijos pablogėjimą; apriboti optinio tankio šlapimo svyravimus; filtravimo ritmo pažeidimas, o tada glomerulų filtracijos sumažėjimas. Etapas lėtinio inkstų nepakankamumo yra diagnozuojama pacientams, buvimo 3-6 mėnesius ar daugiau padidėjęs karbamido serume (daugiau nei 0,35 g / l), sumažintas glomerulų filtraciją iki 25% normalus.
Paveldimo nefrito diferencinė diagnozė turi būti atliekama visų pirma su įgytu glomerulonefritu hematurine forma. Įgijo vis ūmaus glomerulonefrito, prasidedantį 2-3 savaites po ankstesnės infekcijos, extrarenal funkcijos, įskaitant hipertenziją su pirmųjų dienų (paveldimas nefritas, atvirkščiai, hipotenzija), sumažėjo glomerulų filtracijos greitį pradžios, jokių dalinių vamzdinių funkcijų pažeidimas, kadangi kaip ir paveldimiems, jie yra. Gautas glomerulonefritas atsiranda su ryškesniu hematurija ir proteinurija, padidėjusia ESR. Tipiški pokyčiai glomerulų bazės membranoje, būdingi paveldimamam nefritui, yra diagnostiniai.
Diferencinė diagnostika dismetabolinių nefropatija atliekami su lėtiniu inkstų nepakankamumu šeimoje nustatyta kliniškai monotipija inkstų liga, gali svyruoti nuo nefropatijos pielonefrito, kad akmenlige. Vaikai dažnai skundžiasi pilvo skausmu ir periodiškai šlapinasi, šlapimo nuosėdose - oksalatu.
Jei įtariate paveldimą nefritą, pacientą reikia išsiųsti diagnozei paaiškinti specializuotoje nefrologijos skyriuje.
Ką reikia išnagrinėti?
Kaip patikrinti?
Kokie testai reikalingi?
Su kuo susisiekti?
Alporto sindromo gydymas
Režime numatyta riboti didelį fizinį krūvį, likti gryname ore. Dieta yra labai kokybiška, turinti pakankamai aukštos kokybės baltymų, riebalų ir angliavandenių, atsižvelgiant į inkstų funkciją. Labai svarbu identifikuoti ir reabilituoti lėtines infekcijos kamienus. Iš vaistų vartojamas ATP, karokoksilazė, piridoksinas (iki 50 mg per parą), karnitino chloridas. Kursai vyksta 2-3 kartus per metus. Kai hematūrijoje yra skiriama fitoterapija - dilgėlių, dilgėlių, gervuogių uosis, rauguosis.
Užsienio ir vidaus literatūroje yra pranešimų apie gydymą prednizolonu ir citostatikų vartojimą. Tačiau poveikį sunku įvertinti.
Lėtiniu inkstų funkcijos nepakankamumu vartojamas hemodializė ir inkstų transplantacija.
Nėra specifinio (veiksmingo patogenezinio) paveldimo nefrito terapijos metodų. Visos medicininės priemonės yra skirtos inkstų funkcijos sutrikimo prevencijai ir sulėtėjimui.
Riebalai turi būti subalansuoti ir kaloringi, atsižvelgiant į funkcinę inkstų būklę. Jei nėra funkcinės būklės pažeidimų vaiko mityboje, turėtų būti pakankamas baltymų, riebalų ir angliavandenių kiekis. Esant inkstų funkcijos sutrikimo požymiams, baltymų, kalcio ir fosforo angliavandenių kiekis turi būti ribotas, o tai lėtina lėtinio inkstų nepakankamumo vystymąsi.
Fizinis stresas turėtų būti ribotas, vaikams rekomenduojama susilaikyti nuo sporto.
Venkite kontakto su infekciniais pacientais, sumažinkite ūmių kvėpavimo takų infekcijų atsiradimo riziką. Būtina išvalyti lėtinės infekcijos ląsteles. Vaikų, kuriems yra paveldimas nefritas, prevencinės vakcinacijos nėra atliekamos, vakcinacija įmanoma tik pagal epidemiologines nuorodas.
Hormoninis ir imunosupresinis gydymas paveldimiausiu nefritu yra neveiksmingas. Ilgalaikis ciklosporino A ir AKF inhibitorių vartojimas daugelį metų rodo tam tikrą teigiamą poveikį (proteinurijos lygio sumažėjimą ir ligos progresavimo sulėtėjimą).
Gydant pacientus, vartojančius metabolizmą gerinančius vaistus:
- piridoksinas - 2-3 mg / kg per parą 3 dalimis 4 savaites;
- kokarboksilazas - 50 mg į raumenis kas antrą dieną, tik 10-15 injekcijų;
- ATP - 1 ml į raumenis kas antrą dieną, 10-15 injekcijų;
- Vitaminas A - 1000 V / per parą 1 recepcija 2 savaites;
- vitaminas E - 1 mg / kg per parą 1 recepcija 2 savaites.
Toks gydymas gerina bendrą pacientų būklę, mažina vamzdinį disfunkciją ir skiriamas 3 kartus per metus.
Kaip imunomoduliatoriaus galima vartoti levamozolį - 2 mg / kg kūno svorio per dieną 2-3 kartus per savaitę su 3-4 dienų pertraukomis.
Mokslininkams hiperbarinis deguonies kiekis turi teigiamą poveikį hematurijos ir inkstų disfunkcijos sunkumui.
Efektyviausias paveldimojo nefrito gydymo būdas yra skubi inkstų transplantacija. Kai tai nėra pastebėta, kad transplantacijos recidyvo, mažame procentais (apie 5%) gali nefritas plėtrą persodinto inksto, susijęs su antigenų glomerulų pamatinės membranos.
Daug perspektyvių sričių yra prenatalinė diagnozė ir genų inžinerijos terapija. Eksperimentai su gyvūnais rodo didelį efektyvumą perkelti įprastus genus, atsakingus už IV tipo kolageno a-grandinių sintezę į inkstų audinį, po kurio buvo pastebėta normalių kolageno struktūrų sintezė.
Prognozė
Paveldimo nefrito prognozė visada yra rimta.
Prognozuojami nepalankūs paveldimo nefrito srauto kriterijai yra:
- vyrų seksas;
- ankstyvas lėtinio inkstų funkcijos nepakankamumas šeimos nariuose;
- proteinurija (daugiau nei 1 g per parą);
- pagal mikroskopiją glomerulų bazinių membranų storinimas;
- audinio nervo neuritas;
- ištrynimas geno Co4A5.
Geresnės šeimos hematurijos prognozė yra palankesnė.
Использованная литература